Zr、Cr改性CeO2/Al2O3催化劑CO2氧化乙苯脫氫反應研究
2018-07-04 07:31:00冀德坤易玉峰丁福臣劉多強
石油化工 2018年6期
關鍵詞:催化劑
孫 超,冀德坤,易玉峰,丁福臣,劉多強
(1.北京石油化工學院 燃料清潔化及高效催化減排技術北京市重點實驗室,北京 102617;2.中國人民解放軍空軍油料研究所,北京 100076)
苯乙烯是石油化工行業重要的基礎化學品之一。工業生產中,苯乙烯通常是在大量過熱水蒸氣和高溫(高于600 ℃)下由乙苯在鐵鉀催化劑上脫氫制得[1],但工藝存在能耗過高、水蒸氣潛熱難以利用和苯乙烯選擇性低等缺點。因此,研發新型工藝來解決這些問題十分必要。
乙苯氧化脫氫是近些年的研究熱點。大量研究者對O2、SO2、N2、CO2和惰性氣體等氣氛下的乙苯脫氫反應進行了研究,發現O2氣氛下的乙苯氧化脫氫雖然可以降低溫度,但易發生乙苯的深入氧化,產生大量的副產物,反應的選擇性較低[2-3];以SO2為氧化劑會產生多類副產物,且反應過程中還需考慮防腐、污染等問題;N2和He、Ar等惰性氣體氣氛下乙苯轉化率較低[4-6]。CO2氣氛下乙苯脫氫具有加快反應速率、降低能耗、提高苯乙烯選擇性等優點,且對CO2的使用消耗有利于降低溫室效應等不利影響。因此CO2氣氛下的乙苯脫氫工藝是一種綠色環保、節能高效的工藝[7]。
在CO2氣氛下的乙苯脫氫反應中,傳統催化劑催化效果不好且易失活。CeO2是一種相對便宜且廣泛使用的稀土氧化物。Ce具有獨特的f電子結構,使CeO2比其他稀土氧化物具有更好的性能[8]。CeO2在催化脫氫方面的優勢在于其較強的儲放氧能力,而由于純CeO2高溫易燒結、比表面積低等性質[9],CeO2難以直接作為乙苯脫氫催化劑使用,通常加入其他金屬氧化物一起使用。
本工作采用沉淀法制備了Zr、Cr改性的CeO2/Al2O3催化劑,利用BET,XRD,SEM,H2-TPR等技術手段對催化劑進行了表征,考察了Zr、Cr改性后催化劑在CO2氣氛下的乙苯脫氫性能。
1 實驗部分
1.1 催化劑的制備
采用沉淀法制備催化劑。先稱取一定量的Ce(NO3)3·6H2O,Zr(NO3)4·5H2O,Cr(NO3)3·9H2O,γ-A12O3于燒杯中,加入適量蒸餾水,攪拌30 min;然后滴如NH3·H2O至pH為10左右,繼續攪拌2 h,靜置12 h后,真空抽濾得沉淀物,用蒸餾水洗滌沉淀物3次,置于干燥箱中于120 ℃下干燥6 h,最后在550 ℃下焙燒5 h,得催化劑。分別標記為 Ce15Al,Ce15ZrxAl,Ce15CrxAl(x% 為ZrO2或Cr2O3的質量分數)。
1.2 催化劑的表征
BET在北京金埃譜科技有限公司V-Sorb 2800TP型比表面積及孔徑分析儀上進行。取150 mg試樣在200 ℃下預處理120 min,在77 K液氮溫度下通過物理吸附測量催化劑的比表面積。
XRD測試在島津公司XRD-7000型X射線衍射儀上進行。Cu Kα,管電壓40 kV,管電流30 kA,掃描速率 5(o)/min,掃描范圍 2θ = 10o~80o。
SEM表征在FEI公司的Quanta 400F型掃描電子顯微鏡上進行。工作電壓30 kV,反應前噴金處理。
H2-TPR表征在浙江泛泰儀器有限公司FINESORB-3010型程序升溫化學吸附儀上進行。取10 mg催化劑置于U形石英管中,以10 ℃/min的升溫速率升溫至400 ℃,在Ar氣氛下對催化劑進行30 min的預處理。降溫后在5%(φ)H2/Ar氣氛下,以10 ℃/min的升溫速率升溫至800 ℃。
1.3 催化劑的活性評價
催化劑的活性評價在天津大學北洋化工實驗設備公司的MR-B-4型催化劑活性評價系統上進行。反應在常壓下進行,反應溫度600 ℃,CO2與乙苯進料摩爾比10∶1,乙苯進料量用平流泵控制,設為0.1 mL/min,CO2體積流量控制為184 mL/min,催化劑裝填量5 g,催化劑尺寸20~40目。
液體產物在福立分析儀器有限公司GC9720型氣相色譜儀上進行,Super-QTMPLOT色譜柱,進樣口溫度200 ℃,柱溫160 ℃,檢測器溫度280 ℃,H2,N2,空氣的體積比為1∶1∶10。轉化率通過歸一化法計算,乙苯轉化率(X)和苯乙烯選擇性(S)分別由式(1)和(2)計算。
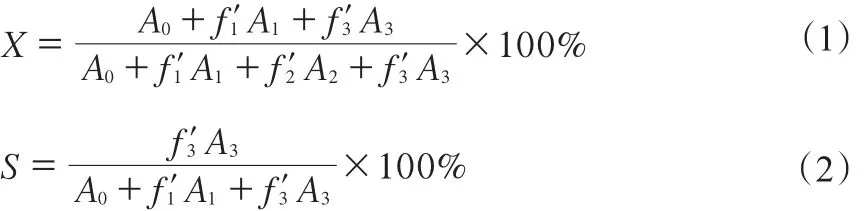
式中,A0,A1,A2,A3分別為苯、甲苯、乙苯和苯乙烯的峰面積;f′1,f′2,f′3分別為甲苯、乙苯和苯乙烯對苯的相對質量校正因子。
2 結果與討論
2.1 催化劑的活性評價
2.1.1 Ce15ZrxAl催化劑的活性
圖1為Zr負載量對CO2氣氛下乙苯脫氫反應中乙苯轉化率和苯乙烯選擇性的影響。由圖1可知,當Zr負載量較低時,隨著Zr負載量的增加,乙苯轉化率提高;當Zr負載量為10%(w)時,乙苯轉化率達最高,在反應12 h時乙苯轉化率達46%,繼續增加Zr負載量,乙苯轉化率下降。隨Zr負載量的增加,苯乙烯選擇性先逐漸增加,在Zr負載量高于12%(w)后選擇性變化較小。
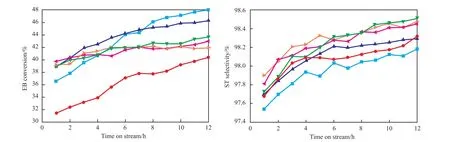
圖1 Ce15ZrxAl催化劑的活性Fig.1 Activities of Ce15ZrxAl catalysts.
2.1.2 Ce15CrxAl催化劑的活性
圖2為Cr負載量對CO2氣氛下的乙苯脫氫反應中乙苯轉化率和苯乙烯選擇性的影響。由圖2可知,當Cr負載量為1%~2%(w)時,催化劑的轉化率較低;當負載量大于3%(w)時,催化劑的初始轉化率大幅提高,乙苯轉化率達55%以上,但轉化率下降較快。添加Cr后,苯乙烯的初始選擇性降低,但隨反應時間的變化幅度較大。綜上可知,當Cr負載量為3%(w)時催化劑具有較高的轉化率和穩定性,從反應初期乙苯轉化率55%到反應12 h后乙苯轉化率48.4%。與Ce15A1催化劑相比,Ce15Cr3A1催化劑的乙苯初始轉化率較高,而苯乙烯初始選擇性較低,穩定性較差。張玉倩[10]對Cr2O3/A12O3催化劑進行了乙苯脫氫實驗,也發現Cr2O3/A12O3催化劑初始活性高,但催化劑活性下降速度快。張學彬[11]將Cr助劑添加到V/Al催化劑中進行乙苯脫氫反應也得到相同結論,添加Cr后催化劑的初始活性增加明顯,但失活速度明顯增加。
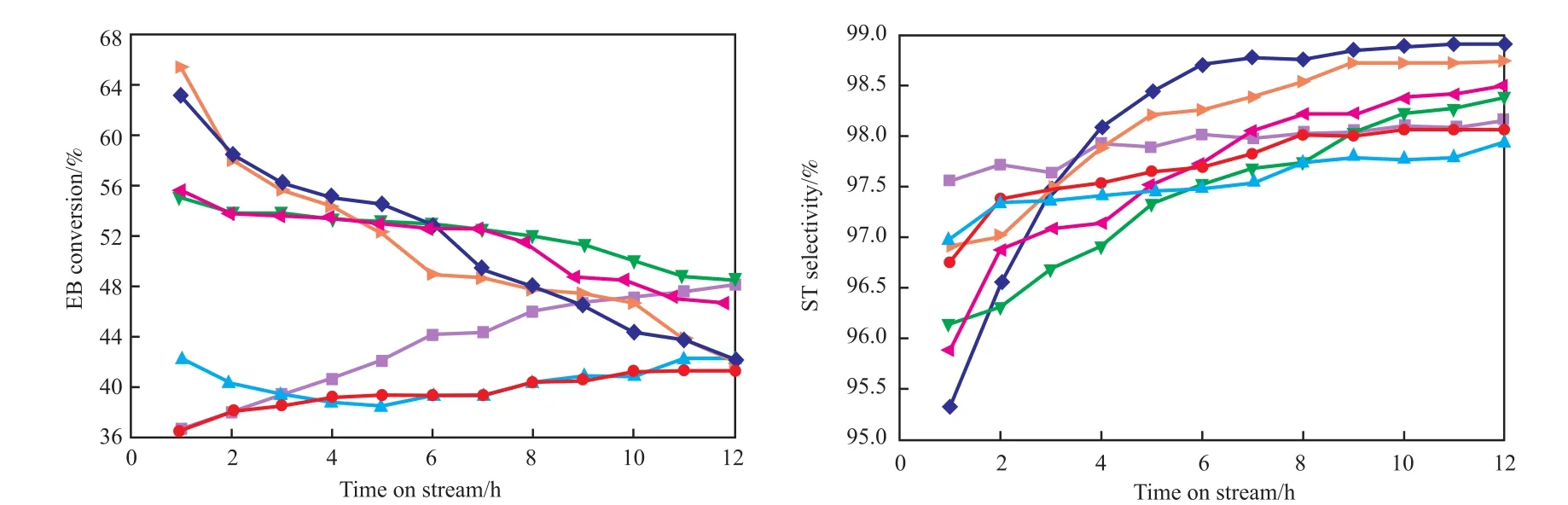
圖2 Ce15CrxAl催化劑的活性Fig.2 Activities of Ce15CrxAl catalysts.
為進一步比較Zr和Cr的加入對催化劑催化性能的影響。對Ce15Al,Ce15Zr10Al,Ce15Cr3Al三種催化劑的活性進行考察。圖3為Ce15Al,Ce15Zr10Al,Ce15Cr3Al三種催化劑反應36 h內乙苯的轉化率和苯乙烯的選擇性。由圖3可知,Ce15Cr3Al催化劑具有較高的乙苯轉化率,在反應前16 h乙苯的轉化率較其他2種催化劑有明顯的優勢,但其乙苯的轉化率下降較快,反應到36 h時乙苯轉化率僅為32.7%,Ce15Al和Ce15Zr10Al催化劑反應36 h后乙苯轉化率分別為35.4%和36.6%,說明Ce15Cr3Al催化劑跟其他兩種催化劑相比在長時間反應中不具備優勢。Ce15Al和Ce15Zr10Al相比,Ce15Zr10Al催化劑的乙苯初始轉化率較好,而Ce15Al催化劑反應初期乙苯的轉化率增長速率快,在8 h后高于Ce15Zr10Al催化劑,但當Ce15Al催化劑的乙苯轉化率在11 h達到最高后,開始下降,且下降速率較快。Ce15Zr10Al催化劑雖然在反應的中期轉化率稍低,但一直較穩定。反應初始階段,Ce15Cr3Al催化劑的苯乙烯選擇性較低,但增長速度快,最終苯乙烯選擇性稍高于其他2種催化劑,且3種催化劑的苯乙烯選擇性均在98.7%以上。綜上可知,Ce15Zr10Al催化劑在長時間反應中具有更好的催化性能。
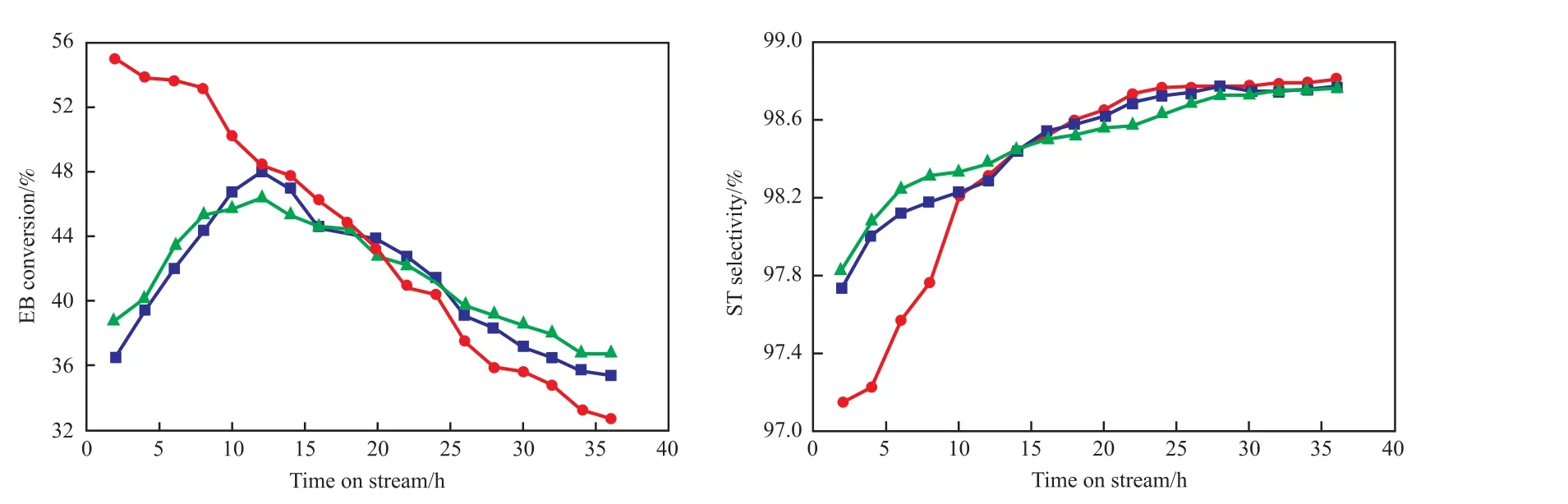
圖3 三種催化劑的活性Fig.3 Activities of three catalysts.
2.2 催化劑的表征
2.2.1 BET和積碳分析結果
表 1為 Ce15Al、Ce15Zr10Al和 Ce15Cr3Al催化劑的BET和積碳表征結果。由表1可看出,在Ce15Al催化劑中摻加Cr和Zr后,催化劑的比表面積均有所降低。但Ce15Zr10Al催化劑的比表面積降幅較小,Ce15Cr3Al催化劑相對另兩種催化劑的比表面積明顯較低。積碳是造成比表面積降低的原因之一,也是導致催化劑失活的重要原因。從反應后積碳量看,反應后3種催化劑均存在明顯的積碳,其中,Ce15Zr10Al催化劑在12 h和36 h反應后積碳量均略低Ce15Al催化劑,但總體相差較小;而Ce15Cr3Al催化劑積碳量遠高于另兩種催化劑,這可能也是Ce15Cr3Al催化劑失活快的重要原因。同時可以發現,反應前12 h積碳較快,隨反應時間的延長,積碳減慢。對于Ce15Al和Ce15Zr10Al催化劑,在12 h反應后均有明顯的積碳和比表面積降低現象,但在反應12 h內乙苯轉化率卻處于持續上升的趨勢,因此,對這兩種催化劑來說,反應初期積碳的存在并沒有造成催化劑的失活,其原因可能在于積碳的存在雖然堵塞了催化劑孔道,造成比表面積降低,但同時積碳也具有一定的催化作用,當積碳量較少時反而對催化劑催化性能有一定的促進作用[12]。Nederlof等[13]的研究表明,反應初期產生的積碳有利于提高乙苯轉化率和苯乙烯選擇性。而當積碳恰好單層覆蓋在催化劑表面時催化劑的活性最佳,隨后積碳繼續沉積,過量的積碳覆蓋催化劑表面、堵塞催化劑的孔道,催化劑的活性開始下降。
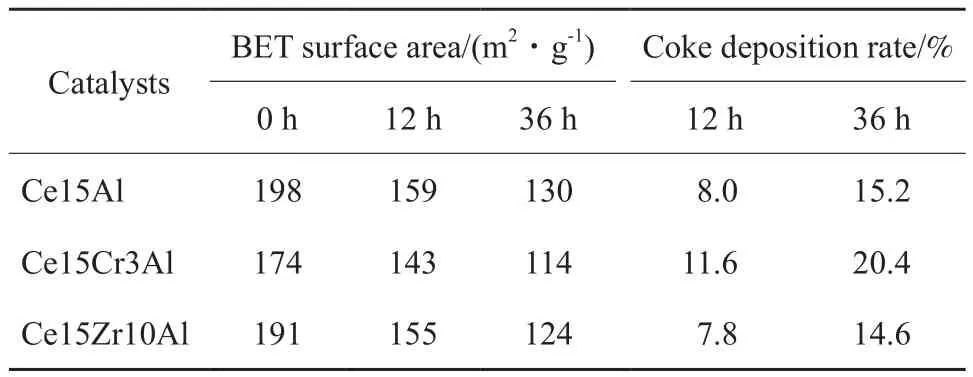
表 1 Ce15Al、Ce15Zr10Al和Ce15Cr3Al催化劑的BET比表面積和積碳率Table 1 BET surface areas and coke deposition amount of Ce15Al,Ce15Zr10Al and Ce15Cr3Al catalysts
2.2.2 XRD分析結果
圖 4為 Ce15Al、Ce15Zr10Al和 Ce15Cr3Al催化劑XRD譜圖。從圖4可看出,對于Ce15Al催化劑,在2θ=36.8o,67.1o處檢測到γ-Al2O3的衍射峰,在 2θ=28.6o,33.1o,47.6o,56.3o處檢測到 CeO2的衍射峰,分別對應于CeO2的(111)、(200)、(220)、(311)立方螢石結構晶相[14]。沒有觀察到明顯的Ce的其他氧化物的衍射峰,說明催化劑中活性組分為立方螢石結構的CeO2。同時也沒有觀察到CeO2和A12O3相互作用物質的峰。有研究表明,600 K以下的焙燒溫度不利于形成低價Ce的氧化物 CeOx和 CeAlO3[15],673~973 K 的焙燒溫度有利于形成非化學計量的CeOx,800 K以上焙燒溫度有利于形成CeAlO3。加入Zr和Cr后并沒有觀察到明顯的ZrO2和Cr2O3衍射峰。加入Zr后,催化劑中A12O3的衍射峰強度減小,說明Zr的存在增強了A12O3的分散性,同時衍射峰向高角度移動,這證明了CeO2-ZrO2固溶體的存在,衍射峰角度的移動是因為晶格中Ce4+部分被Zr4+取代,由于Zr4+的離子半徑小于Ce4+的離子半徑,因此Zr4+進入CeO2晶格中引起晶格收縮,晶格參數變小,使得衍射峰角度向高角度移動[16-17]。同時,催化劑的衍射峰峰形更加尖銳,說明加入Zr后催化劑的晶粒尺寸變大。加入Cr后,催化劑的衍射峰位置沒有發生明顯變化,催化劑衍射峰峰形變得尖銳,說明催化劑的晶粒變大。葉興南等[18]對Cr2O3/Al2O3催化劑進行XRD分析,發現當Cr2O3含量高于25%(w)時才會出現明顯的Cr2O3的衍射峰。
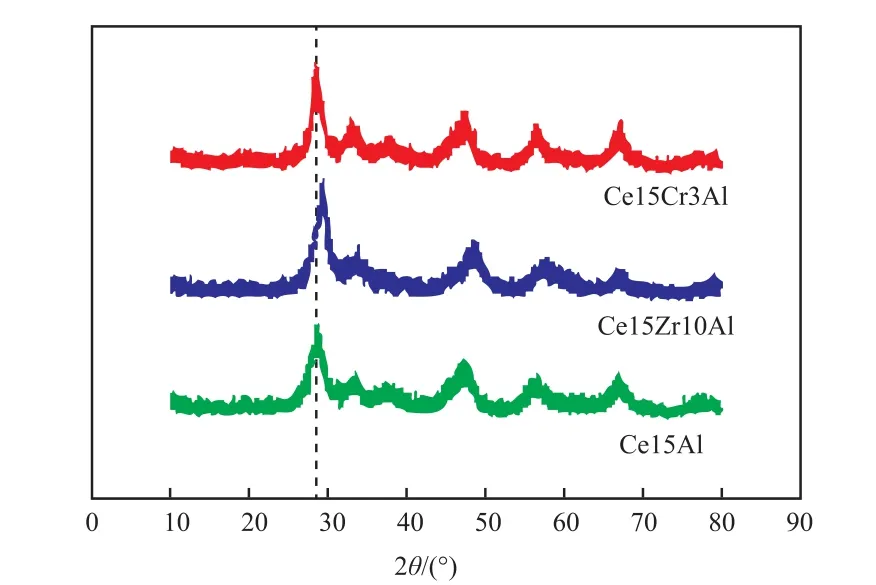
圖4 催化劑的XRD譜圖Fig.4 XRD patterns of catalysts.
2.2.3 SEM分析結果
圖 5為 Ce15Al、Ce15Zr10Al和 Ce15Cr3Al催化劑的SEM照片。由圖5a可知,Ce15Al催化劑的顆粒尺寸較小且無固定的幾何形態,催化劑存在明顯的燒結現象,部分顆粒凝結在一起,分散性較差。由圖5b可知,加入Zr后,催化劑的形貌有所變化,催化劑的晶粒尺寸整體變大,這與XRD分析結果相一致,催化劑的顆粒尺寸大小不一,顆粒間分散性較好,催化劑的燒結現象明顯改善,說明Zr的加入增加了催化劑的抗燒結能力。而從圖5c可知,加入Cr后,催化劑的顆粒粒徑明顯增大,且存在較嚴重的團聚現象,顆粒分散不均勻,這可能也是造成Ce15Cr3Al催化劑的比表面積較低的重要原因。

圖5 催化劑的SEM照片Fig.5 SEM images of catalysts.
2.2.4 H2-TPR分析結果
圖 6為 Ce15Al、Ce15Zr10Al和 Ce15Cr3Al催化劑的H2-TPR譜圖。由圖6可知,Ce15Al催化劑的H2-TPR譜圖中在479 ℃和697 ℃處觀察到2個還原峰,分別對應于表面Ce4+的還原和體相Ce4+的還原[19]。在催化劑中加入Zr后沒有出現新的衍射峰,這是因為Zr4+難以被還原,同時催化劑表面Ce4+的還原峰強度有所減弱,體相Ce4+的還原峰變得不明顯,催化劑的還原性有所降低。這可能也是反應中期Ce15Zr10Al催化活性稍差的原因。Zhang等[17]認為加入Zr后,催化劑的體相晶格氧遷移率降低,這造成了體相Ce4+的還原峰消失,還原性降低,但同時這也是加入Zr后催化劑的熱穩定性變好的原因。催化劑中加入Cr后,催化劑的H2-TPR譜圖有明顯變化,分別在373,477,672 ℃處觀察到3個明顯的還原峰,分別對應于Cr6+向 Cr3+的還原[20]、表相 Ce4+的還原和體相Ce4+的還原,還原峰面積明顯增加,Ce4+還原溫度降低,催化劑的還原性增強,這可能也是反應初始階段Ce15Cr3Al催化劑催化效果好的原因之一。
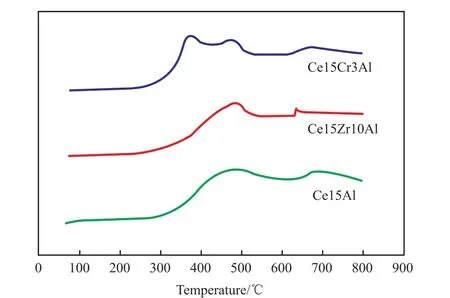
圖6 催化劑的H2-TPR譜圖Fig.6 H2-TPR spectra of the catalysts.
3 結論
1)Cr改性Ce15Al催化劑,Cr最佳負載量為3%(w),改性后,反應初期乙苯轉化率55%,反應12 h后乙苯轉化率48.4%;Zr改性Ce15Al催化劑,Zr最佳負載量為10%(w),反應12 h時乙苯轉化率達46%。Zr改性的催化劑在長時間的反應中具有更好的催化性能。
2) Cr改性后,Ce15Al催化劑的比表面積明顯降低,團聚現象嚴重;反應后催化劑的積碳率較高,催化劑失活較快。Zr改性后,Zr進入Ce晶格中形成CeO2-ZrO2固溶體,改善了Ce15Al催化劑的燒結現象,催化劑的比表面積和還原性稍有降低,但熱穩定性增強,對抑制積碳有積極作用。
[1] 熊麗萍,李國范,譚忠雋,等. 乙苯脫氫制苯乙烯催化劑的研究進展[J].石油化工,2015,44(4):517-522.
[2] 蔡衛權,李會泉,張懿. CO2選擇性氧化乙苯制苯乙烯[J].化學進展,2004,16(3):406-413.
[3] Kustrowski P,Segura Y,Chmielarz L,et al. VOxsupported SBA-15 catalysts for the oxidative dehydrogenation of ethylbenzene to styrene in the presence of N2O[J].Catal Today,2006,114(2/3):307-313.
[4] Nederlof C,Talay G,Kapteijn F,et al. The role of RWGS in the dehydrogenation of ethylbenzene to styrene in CO2[J].Appl Catal,A,2012,423/424(8):59-68.
[5] Saito K,Okuda K,Ikenaga N,et al. Role of lattice oxygen of metal oxides in the dehydrogenation of ethylbenzene under a carbon dioxide atmosphere[J].J Phys Chem A,2010,114(11):3845-3854.
[6] Balasamy R J,Tope B B,Khurshid A,et al. Ethylbenzene dehydrogenation over FeOx/(Mg,Zn)(Al)O catalysts derived from hydrotalcites:Role of MgO as basic sites[J].Appl Catal,A,2011,398(1/2):113-122.
[7] 王維維,韓武剛,陳樹偉,等. VOx/SBA-16催化CO2氧化乙苯脫氫性能[J].應用化工,2017,46(12):2306-2309.
[8] Xu Jie,Wang Luncun,Liu Yongmei,et al. Mesostructured CeO2as an effective catalyst for styrene synthesis by oxidative dehydrogenation of ethylbenzene[J].Catal Lett,2009,133(3/4):307-313.
[9] 張棲,張明,付名利,等. Al2O3對CexZr1-xO2氧化還原性能的影響[J].中國科技論文在線,2010,5(6):448-452.
[10] 張玉倩. 乙苯CO2脫氫催化劑穩定性的研究[D].上海:華東理工大學,2011.
[11] 張學彬. 釩系催化劑在乙苯混合脫氫過程中的應用[D].西安:陜西師范大學,2008.
[12] Pereira M F R,Orfao J J M,Figueiredo J L. Oxidative dehydrogenation of ethylbenzene on activated carbon fi bers[J].Carbon,2002,40 (13):2393-2401.
[13] Nederlof C,Kapteijn F,Makkee M. Catalysed ethylbenzene dehydrogenation in CO2or N2—Carbon deposits as the active phase[J].Appl Catal,A,2012,417/418(1):163-173.
[14] Rao R,Zhang Qingyun,Liu Huade,et al. Enhanced catalytic performance of CeO2conf i ned inside carbon nanotubes for dehydrogenation of ethylbenzene in the presence of CO2[J].J Mol Catal A:Chem,2012,363/364(11):283-290.
[15] Reddy B M,Rao K N,Reddy G K,et al. Structural characterization and oxidehydrogenation activity of CeO2/Al2O3and V2O5/CeO2/Al2O3catalysts[J].J Phys Chem C,2007,111(50):18751-18758.
[16] 金銀龍,王國清,張利軍,等. 焙燒溫度對鈰鋯固溶體性能的影響[J].石油化工,2017,46(7):857-861.
[17] Zhang Guizhen,Zhao Zhen,Xu Junfeng,et al. Comparative study on the preparation,characterization and catalytic performances of 3DOM Ce-based materials for the combustion of diesel soot[J].Appl Catal,B,2011,107(3/4):302-315.
[18] 葉興南,華偉明,樂英紅,等. CO2氣氛下負載型Cr2O3催化劑上乙苯脫氫制苯乙烯反應[J].催化學報,2004,25(7):581-585.
[19] Li Xiaohong,Feng Jie,Fan Hongxia,et al. The dehydrogenation of ethylbenzene with CO2over CexZr1-xO2solid solutions[J].Catal Commun,2015,59(1):104-107.
[20] 李惠云,樂英紅,繆長喜,等. 溶膠-凝膠法制備用于CO2氣氛下乙苯脫氫反應的高活性Cr2O3-SiO2催化劑[J].催化學報,2006,27(1):4-6.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
