黃原膠和瓜爾豆膠復配比例對復原乳穩定性的影響
2018-08-09 01:57:40文/劉丹
中國乳業 2018年7期
文/劉 丹
(廣東燕塘乳業股份有限公司)
復原乳是一種具有膠體特性的多分散體系,主要由蛋白質、脂肪等成分組成,貯藏過程中易出現絮凝、分層、蛋白質沉淀和脂肪上浮等不穩定現象,嚴重影響品質。如何提高其穩定性是復原乳生產企業急需解決的問題。本文圍繞改善復原乳穩定性和口感2 個關鍵指標,研究了多糖類乳化穩定劑(黃原膠和瓜爾豆膠復配)對復原乳穩定性的影響,并探討其作用機理,為制備穩定性較好的復原乳提供理論支撐和方法指導。
1 材料與方法
1.1 材料與儀器
材料:全脂奶粉;瓜爾豆膠、黃原膠(食品級);白砂糖;油紅O(分析純),甲醇、乙腈、異丙醇、三氯甲烷(色譜級);疊氮鈉(分析純);飲用水。
儀器:高速分散剪切機;高壓均質機;Mastersizer 2000型激光粒度分析儀;Waters 600高效液相色譜儀;3000型蒸發光散射檢測器;754型分光光度計;BHW-IV型電熱恒溫水槽;分析天平;離心機。
1.2 試驗方法
1.2.1 復原乳的制備
復原乳的制備配方見表1,工藝流程如圖1所示。將主、輔原料混合均勻,在勻速攪拌狀態下將其緩慢加入水中,并于60 ℃中保溫30 min,充分水化;水化結束后,20 MPa下均質;隨后灌裝和滅菌,滅菌條件為121 ℃、15 min;最后迅速冷卻至常溫。
1.2.2 界面蛋白濃度的測定
取一定量的復原乳,10 000 r/min、4 ℃條件下離心30 min。離心完成后將樣品分離成乳析層和下層清液層,轉移下層的清液,采用凱氏定氮法測定下層清液中的蛋白質含量(N×5.3)[1]。
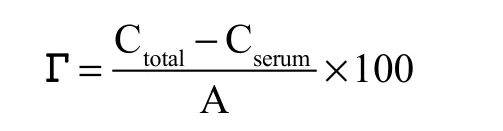
式中:Г為界面蛋白濃度(mg/m2),Ctotal為復原乳中蛋白質濃度(g/g),Cserum為下層清液中蛋白質濃度(g/g),A為液滴的比表面積(m2/g)。
1.2.3 粒度分布的測定
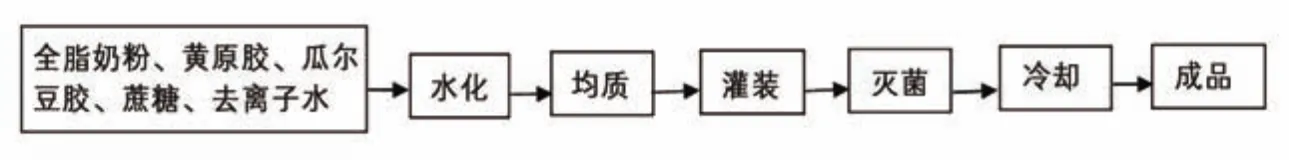
圖1 復原乳制備的基本工藝流程
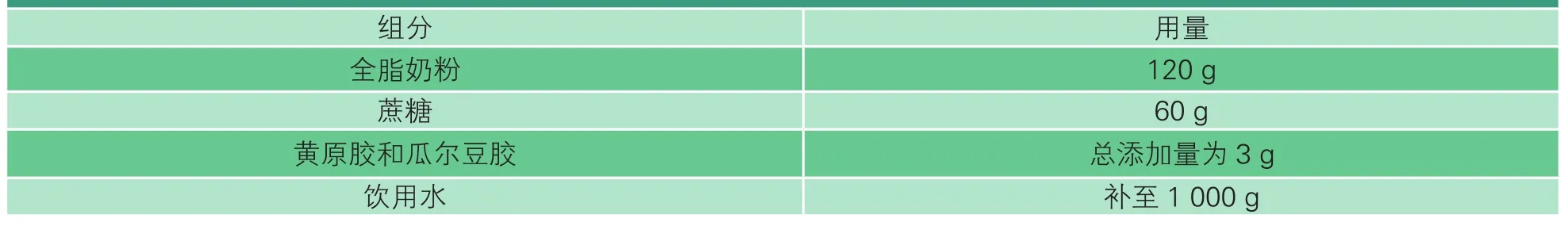
表1 復原乳的制備配方

表2 Mastersizer 2000型激光粒度分析儀參數
復原乳樣品按1∶1000(w/w)?的比例用去離子水稀釋,然后用Mastersizer 2000型激光粒度分析儀測定粒度分布。參數設定見表2[2]。
體積平均直粒徑d4,3計算公式為:
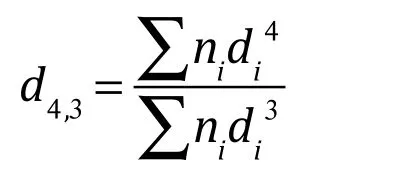
其中,ni為直徑為di的脂肪球的數量。
1.2.4 脂肪部分聚結率的測定
準確稱取0.015 g油紅O色素,加入到1 000 g粟米油中混合,常溫下慢速攪拌約12 h使油紅O充分溶解,避光保存。準確稱取20 g樣品和10 g油紅O溶液置于50 mL離心管中,充分混合均勻,10 000 r/min、25 ℃條件下離心30 min,移取上層澄清透明的紅色油液倒入比色杯,520 nm波長下測定吸光度值[3]。計算公式為:
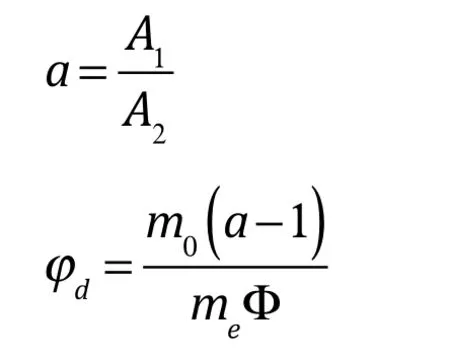
式中:A1為離心前油紅O溶液的吸光度值,A2為離心后油紅O溶液的吸光度值,φd為脂肪部分聚結率,m0為樣品中加入的油紅O溶液質量,me為樣品質量,Φ為樣品中脂肪的質量分數。
1.2.5 離心乳析率和沉淀率的測定
取10 g復原乳樣品于離心管中,3 000 r/min、25 ℃條件下離心20 min,準確稱量上部油脂部分的質量(m1);棄去上部溶液,準確稱取沉淀物的質量(m2),計算離心乳析率和離心沉淀率,計算公式為:

2 結果與分析
2.1 乳化劑不同配比對復原乳界面蛋白濃度的影響
界面蛋白濃度(Г)表示的是液滴表面單位面積上的蛋白質含量,是反映乳濁液凝結穩定性的一個重要參數,不僅與界面蛋白質含量(PA)有關,還取決于乳濁液粒徑的大小。由圖2可知,在單獨使用黃原膠或者瓜爾豆膠的情況下,蛋白質—黃原膠體系的界面蛋白濃度(Г)比蛋白質—瓜爾豆膠體系的Г值高,這可能是因為瓜爾豆膠和黃原膠屬于非吸附多糖,不與蛋白質產生競爭性吸附,只通過靜電吸附、疏水作用或者氫鍵作用與蛋白質形成復合物。中性條件下,蛋白質和黃原膠都帶負電,而瓜爾豆膠不帶電,黃原膠可以與蛋白質的部分帶正電區域發生靜電吸引作用,因此蛋白質—黃原膠體系的Г值較高[4]。
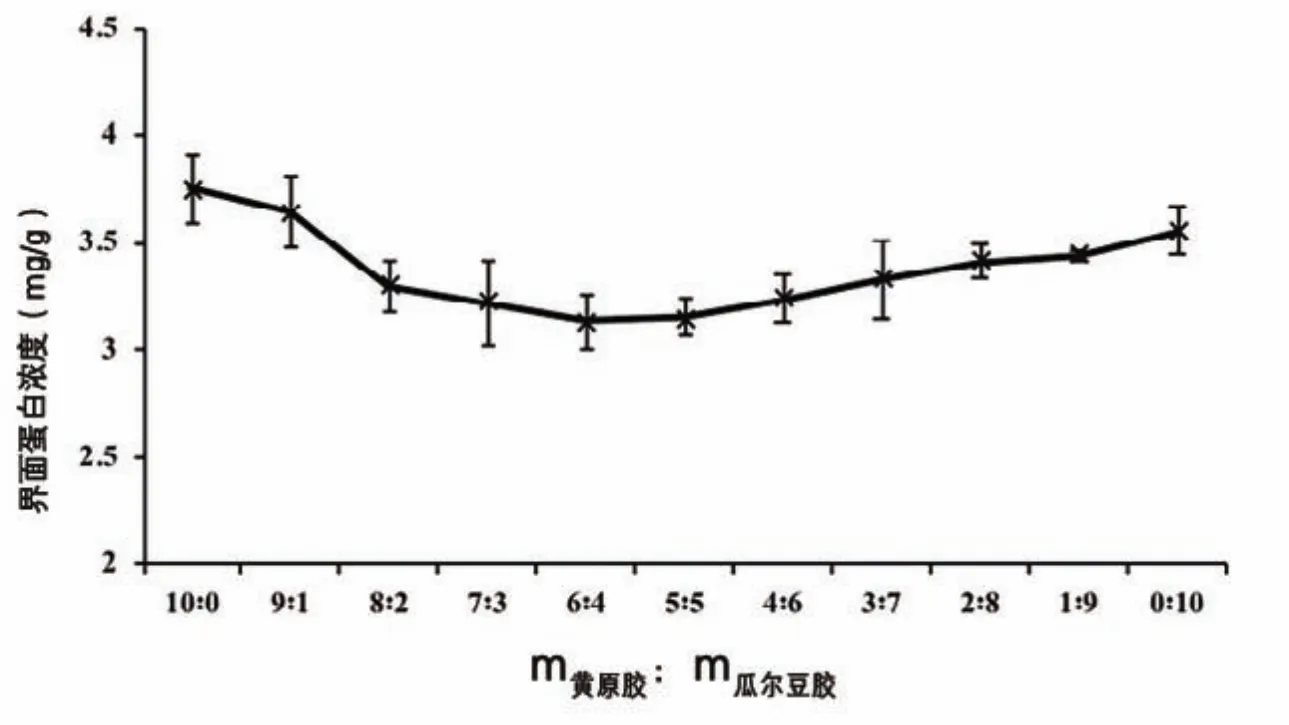
圖2 乳化劑不同配比對復原乳液界面蛋白濃度的影響
當黃原膠、瓜爾豆膠兩者復配使用時,隨著黃原膠的減少,Г值呈現先降低后穩定再升高的趨勢。這是由于黃原膠濃度降低,使得黃原膠與蛋白質部分帶正電區域發生的靜電吸引作用減小,Г值呈現明顯降低。隨著瓜爾豆膠的增加,黃原膠和瓜爾豆膠的共同作用導致Г值變化不明顯。當m黃原膠∶m瓜爾豆膠小于4∶6時,黃原膠與蛋白質的靜電作用明顯減弱,瓜爾豆膠與蛋白質的相互作用占主導地位,Г值增加。
2.2 乳化劑不同配比對復原乳粒度分布的影響
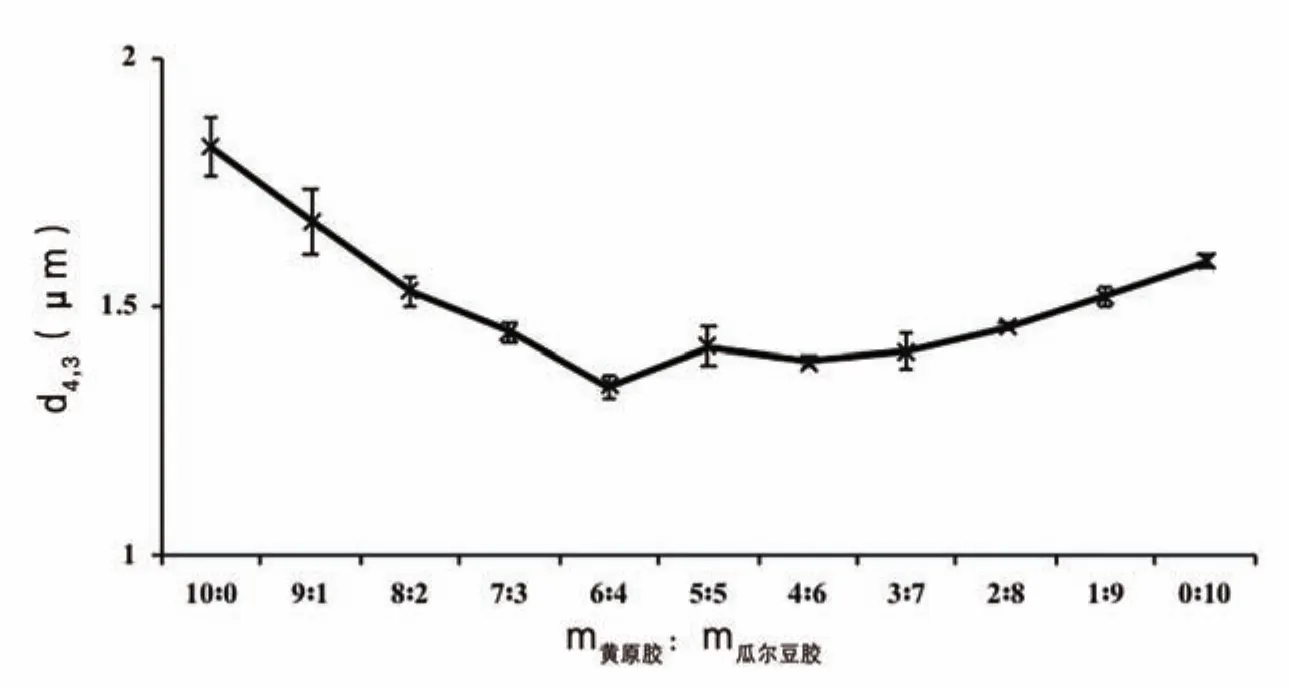
圖3 乳化劑不同配比對復原乳粒度分布的影響
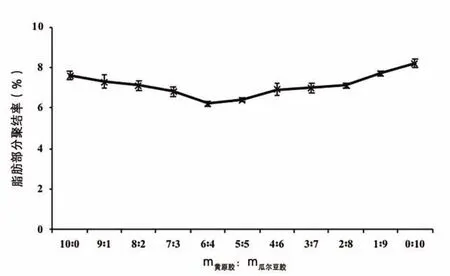
圖4 乳化劑不同配比對復原乳脂肪部分聚結率的影響
由圖3可知,乳化劑不同配比對復原乳粒度分布的影響與其對界面蛋白濃度的影響趨勢一致。如前所述,界面蛋白濃度不僅與界面蛋白質含量有關,還取決于乳濁液的粒徑大小,而粒徑大小與體系黏度有關。當體系黏度大時,復原乳的體積平均直粒徑(d4,3)值偏大。相同濃度下,黃原膠溶液的黏度比瓜爾豆膠溶液高,所以蛋白質—黃原膠復合體系的d4,3值大于蛋白質—瓜爾豆膠復合體系的d4,3值。當黃原膠、瓜爾豆膠兩者復配使用時,隨著黃原膠的減少,d4,3值呈現先降低后穩定再升高的趨勢。原因可能是,一方面黃原膠減少時靜電吸附的蛋白質數量減少,另一方面體系中未吸附的黃原膠減少,降低了未吸附黃原膠與蛋白質熱力學不相容性,從而減小了體系發生排斥絮凝的機率,抑制了液滴聚集,所以d4,3值降低[5]。由于2 種乳化劑和蛋白質作用方式不同,當m黃原膠∶m瓜爾豆膠為5∶5、4∶6和3∶7時,2 種相互作用方式共同維持了復原乳體系的穩定性。當m黃原膠∶m瓜爾豆膠小于3∶7時,黃原膠維持體系穩定性的作用減弱,瓜爾豆膠濃度較高,界面蛋白質吸附量大,蛋白質分子在界面上沒有足夠空間展開,結構被壓縮,瓜爾豆膠與蛋白質間的相互作用受到抑制,容易引起體系排斥絮凝的發生,因此粒徑反而增大。
2.3 乳化劑不同配比對復原乳脂肪部分聚結率的影響
對乳濁液的脂肪部分聚結率進行檢測,結果如圖4所示。單獨使用黃原膠的脂肪聚結率比單獨使用瓜爾豆膠的脂肪聚結率低,這是由于在相同條件下,蛋白質—黃原膠復合體系界面膜的黏彈性特征比蛋白質—瓜爾豆膠復合體系界面膜的黏彈性特征強,更易于降低兩相的界面張力,阻止脂肪球互相聚結[6]。當m黃原膠∶m瓜爾豆膠為10∶0~6∶4時,隨著黃原膠用量減少,脂肪部分聚結率下降;當m黃原膠∶m瓜爾豆膠小于5∶5時,脂肪部分聚結率緩慢上升。原因可能是,當黃原膠用量減少、瓜爾豆膠用量增加時,體系中未吸附的黃原膠減少,同時瓜爾豆膠又以不同于黃原膠與蛋白質作用的方式維持體系的穩定性,減小了體系發生排斥絮凝的幾率,抑制了脂肪球互相聚集,脂肪部分聚結率隨之下降。當黃原膠的用量減少到一定值時,黃原膠與蛋白質的靜電吸附作用很弱,蛋白質從界面解析的數量超過了一定范圍,降低了界面膜的黏彈性,而增加的瓜爾豆膠又不能補充界面膜損失的粘彈性,導致體系的脂肪部分聚結率升高。
2.4 乳化劑不同配比對復原乳離心乳析率和離心沉淀率的影響
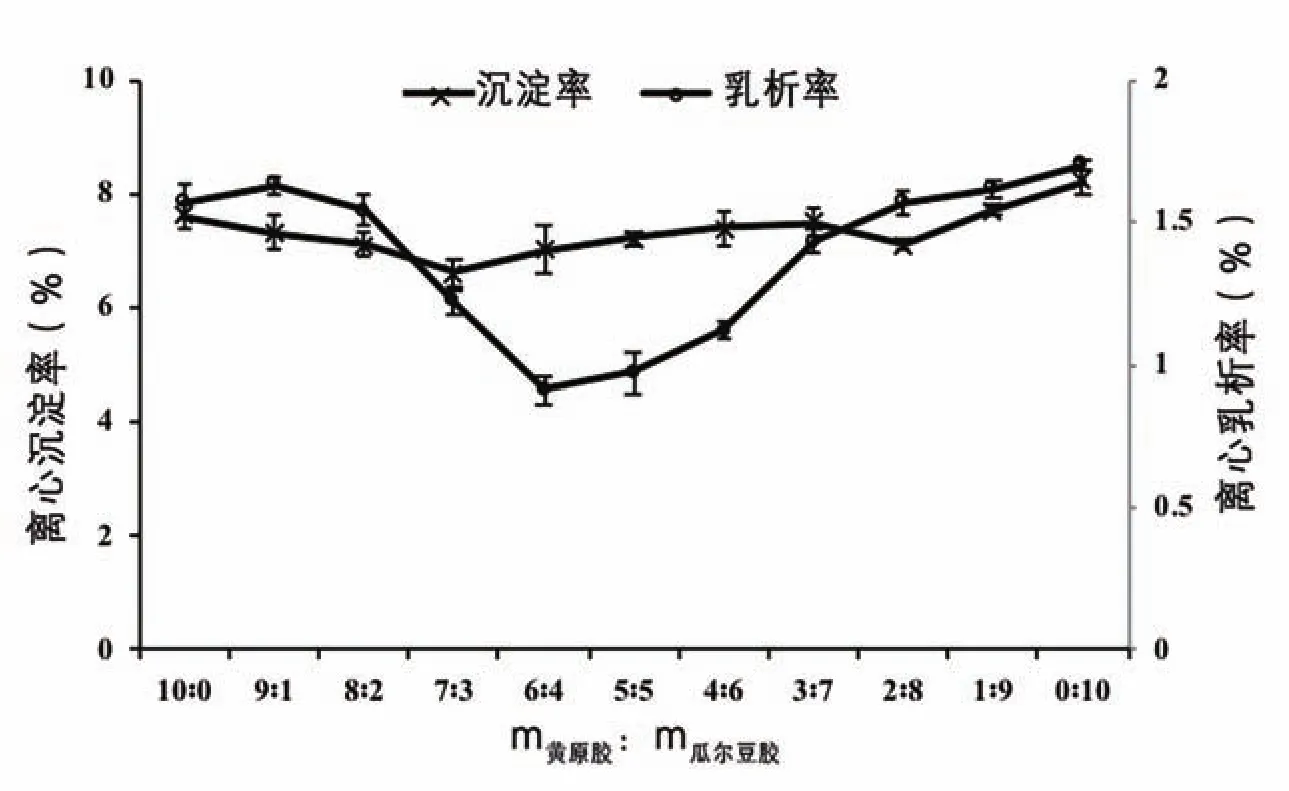
圖5 乳化劑不同配比對復原乳離心乳析率和離心沉淀率的影響
體系離心乳析率和離心沉淀率反映了體系的穩定性狀況。由圖5可知,當m黃原膠∶m瓜爾豆膠為10∶0~7∶3時,隨著黃原膠用量減少,離心沉淀率下降,當m黃原膠∶m瓜爾豆膠小于7∶3時,離心沉淀率緩慢上升;當m黃原膠∶m瓜爾豆膠為10∶0~6∶4時,隨著黃原膠用量減少,離心乳析率下降,當m黃原膠∶m瓜爾豆膠小于6∶4時,離心乳析率上升。這一結果說明,隨著黃原膠用量減少,體系的穩定性先增加后減弱,這與上述結果討論一致,即適度的m黃原膠∶m瓜爾豆膠能減少蛋白質間的聚集,從而提高復原乳的穩定性,但單純某種乳化劑占比過高時,乳化劑與蛋白質的作用容易達到過飽和狀態,過量的蛋白質漸漸從油相中分離沉淀出來,破壞復原乳的穩定性。
2.5 乳化劑不同配比對貯藏期間復原乳感官評分的影響
依據液態乳的感官評定標準NY 5140-2005[7]對復原乳進行感官評分,結果見表3。由表3可以看出,隨著貯藏時間的延長,各配比下復原乳的得分均有所下降,其中m黃原膠∶m瓜爾豆膠為6∶4時感官得分最高且分數下降幅度最小。添加適量乳化劑能在一定程度上改善復原乳口感稀薄、有稍許澀味存在等問題,但是如果某一種乳化劑添加量過多,可能會將其自身的缺點體現在復原乳上。復原乳貯藏期間,由于脂肪球的聚結上浮和蛋白質的聚集沉淀,一方面導致復原乳出現分層現象,嚴重時該現象不可逆,影響復原乳的外觀;另一方面減少了復原乳的香氣成分,影響復原乳的滋味與氣味。綜合考慮,m黃原膠∶m瓜爾豆膠為6∶4時復原乳穩定性最好,感官品質最佳。
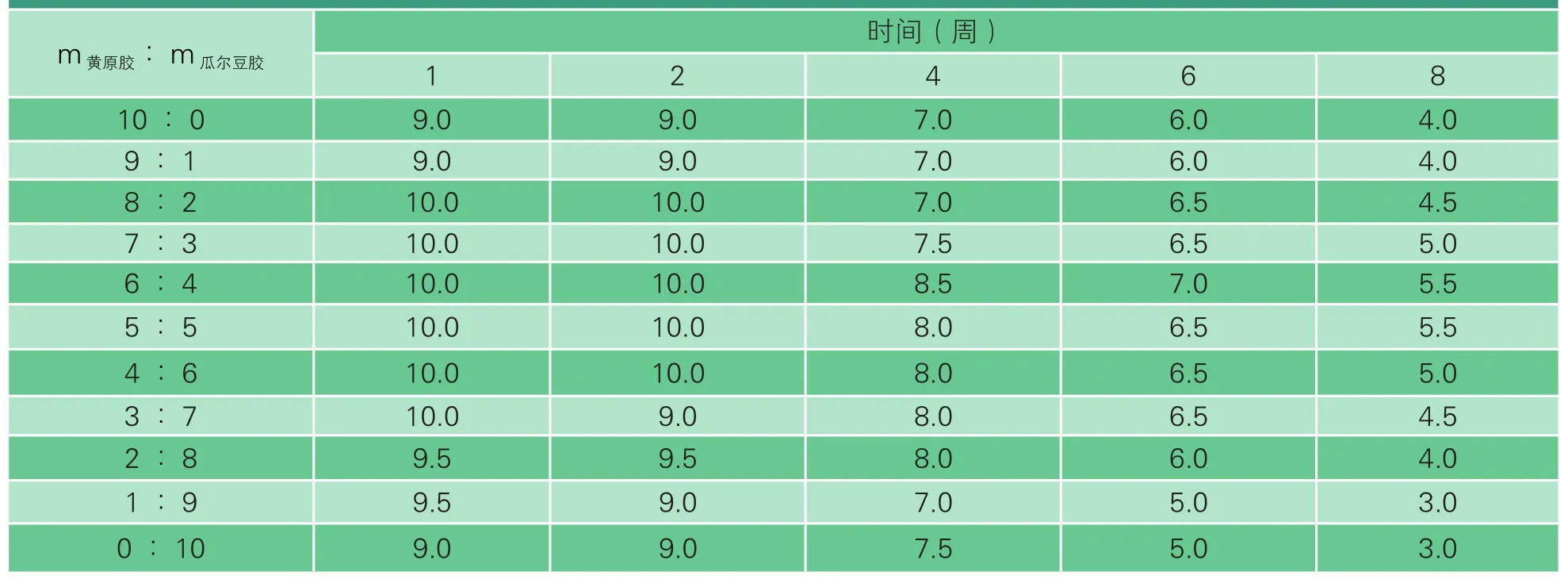
表3 乳化劑不同配比時貯藏期間復原乳的感官評分
3 結論
乳化劑總用量不變,復原乳的界面蛋白濃度、粒徑、脂肪部分聚結率在黃原膠和瓜爾豆膠的復配比例為10∶0~6∶4范圍時呈下降趨勢,在6∶4~0∶10范圍時呈上升或緩慢上升趨勢。復原乳離心乳析率在黃原膠和瓜爾豆膠的復配比例為10∶0~6∶4范圍時呈下降趨勢,在6∶4~0∶10范圍時呈上升趨勢;離心沉淀率則在黃原膠和瓜爾豆膠的復配比例為10∶0~7∶3范圍時呈下降趨勢,在7∶3~0∶10范圍時呈上升趨勢。
隨著貯藏時間的延長,黃原膠和瓜爾豆膠不同配比下復原乳的感官評分出現了明顯差異,感官得分均有所下降,當m黃原膠∶m瓜爾豆膠為6∶4和5∶5時,復原乳貯藏后的感官評分差異較小。C
猜你喜歡
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
當代陜西(2020年13期)2020-08-24 08:22:02
中國外匯(2019年17期)2019-11-16 09:31:14
制造技術與機床(2017年5期)2018-01-19 02:49:17
金秋(2017年4期)2017-06-07 08:22:16
中國材料進展(2016年10期)2016-12-26 06:50:20
濰坊學院學報(2016年2期)2016-12-01 13:00:11
新聞傳播(2015年11期)2015-07-18 11:15:04
現代企業(2015年1期)2015-02-28 18:43:18
新高考·高一物理(2014年1期)2014-09-18 01:26:07
