砂仁藥材的綜合利用試驗研究
2018-09-22 04:15:36張繼斌
農產品加工 2018年17期
關鍵詞:黃酮
張繼斌
(勁牌生物醫藥有限公司,中藥保健食品質量與安全湖北省重點實驗室,湖北大冶 435100)
砂仁為姜科植物陽春砂(Amomum villosum Lo-ur.)、綠殼砂(Amomum villosum Lour.var.xanthioides T.L.Wu et Senjen) 或海南砂 (Amomum longiligularg T.L.wu)的干燥成熟果實,具有化濕開胃、溫脾止瀉、理氣安胎等功能,主要用于濕濁中阻、脘痞不饑、脾胃虛寒、嘔吐泄瀉、妊娠惡阻、胎動不安等[1]。砂仁主要成分為揮發油類成分[2-4],但其中也含有黃酮類成分[5]和多糖類成分[6]。
試驗通過正交試驗法優選藥材粒度、溶劑倍數、浸泡時間、提取時間等影響因素,以提取得到揮發油中乙酸龍腦酯、提取物中總黃酮和總多糖提取率為考查指標,確定了砂仁藥材綜合利用的最佳工藝條件。通過對砂仁藥材進行一次提取,同時得到砂仁揮發油、砂仁黃酮、砂仁多糖提取物,對砂仁藥材進行了綜合利用,具有顯著的經濟效益和社會效益。
1 試驗材料
1.1 儀器與設備
6890型氣相色譜儀,安捷倫科技公司產品;TU1901型紫外可見分光光度計,北京普析通用有限公司產品;AB135-S型電子天平,梅特勒-托利多公司產品;FA2004型電子天平,上海精密科學儀器有限公司產品;SK8200LHC型超聲提取器,上海科導超聲儀器有限公司產品。
1.2 材料與試劑
砂仁藥材,由勁牌生物醫藥有限公司提供;乙酸龍腦酯對照品、蘆丁對照品、葡萄糖對照品,由中國食品藥品監督檢定研究院提供;苯酚、濃硫酸、甲醇、乙醇、亞硝酸鈉、硝酸鋁、氫氧化鈉,均為分析純;純凈水。
2 試驗方法
2.1 乙酸龍腦酯含量測定
按《中國藥典》2015版一部“砂仁”項下規定的方法測定。
2.2 總黃酮含量測定
2.2.1 對照品溶液的制備
精密稱取5 mg蘆丁標準品,用30%乙醇溶液溶解并定容至50 mL,作為對照品溶液。
2.2.2 標準曲線的制備
精密量取對照品溶液0.2,0.4,0.6,0.8,1.0 mL,置于10 mL容量瓶中,然后加入0.3 mL 5%NaNO2溶液,混勻,放置6 min;加入0.3 mL 10%AL(NO3)3溶液,混勻,放置6 min;然后加入1 mol/L的NaOH溶液2 mL,混勻,放置15 min;用30%乙醇定容至10 mL,搖勻。以相應試劑為空白,于波長510 nm處測定吸光度,以吸光度為縱坐標、質量濃度為橫坐標繪制標準曲線。
2.2.3 供試品溶液的制備
稱取適量的提取物粉末樣品,加入適量的50%乙醇至50 mL容量瓶中,搖勻,超聲溶解,定容,放置,作為供試品溶液。
2.2.4 含量測定
精密吸取供試品溶液1.0 mL,置于10 mL容量瓶中,按照2.2.2的方法,自“加入0.3 mL 5%NaNO2溶液”起,依法測定吸光度,計算即得。
2.3 總多糖含量測定
2.3.1 對照品溶液的制備
精確稱取105℃干燥至恒質量的葡萄糖對照品50 mg,置于50 mL容量瓶中,加水溶解并稀釋至刻度。再量取1 mL,置于50 mL容量瓶中,加水稀釋至刻度。
2.3.2 標準曲線的制備
吸取葡萄糖標準液0.2,0.4,0.6,0.8,1.0 mL,分別置于具塞比色管中,分別加水補至2.0 mL,分別精密加入5%苯酚溶液1 mL,搖勻,迅速精密加入濃硫酸5 mL,搖勻,置沸水浴中煮沸15 min,取出冷卻至室溫,以相應試劑為空白,于波長490 nm處測定吸光度,以吸光度為縱坐標、質量濃度為橫坐標繪制標準曲線。
2.3.3 供試品溶液的制備
稱取樣品約15 mg,于50 mL容量瓶中,加入40 mL水,超聲溶解、定容、過濾,作為供試品溶液備用。
2.3.4 顯色測定
取上述供試品溶液1 mL,加水至2.0 mL,按照2.3.2的方法,自“精密加入5%苯酚溶液1 mL”起,依法測定吸光度,從標準曲線上讀出供試品溶液中葡萄糖的質量濃度,計算即得。
3 正交試驗
3.1 因素水平設計
采用水加熱提取,其影響因素主要有藥材粒度、溶劑倍數、浸泡時間、提取時間[7],采用L9(34)正交表優化提取條件,考查上述4個因素和3個水平。
因素與水平設計見表1。
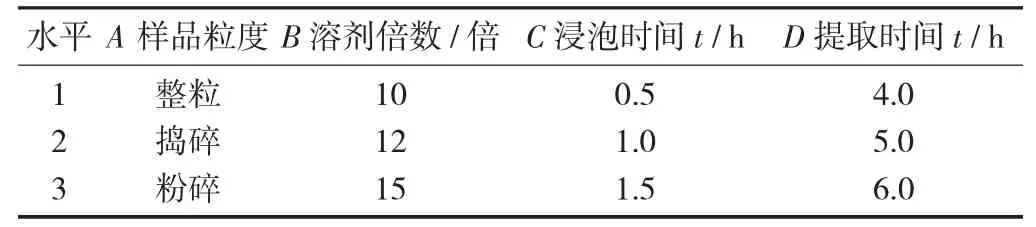
表1 因素與水平設計
3.2 樣品制備
將砂仁藥材分別按要求稱取質量為100 g的樣品9份,按正交試驗表的條件分別進行加熱提取,收集得到揮發油,準確測量其體積。
揮發油相關數據見表2。
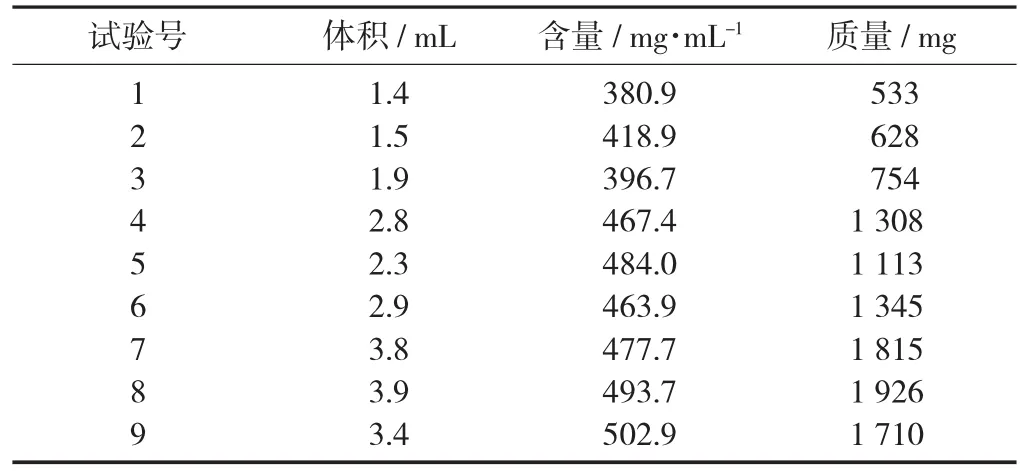
表2 揮發油相關數據
3.3 含量測定
按試驗方法測定揮發油中的乙酸龍腦酯的含量,并分別計算質量。結果如表2所示。
3.4 正交分析
以揮發油中乙酸龍腦酯質量為考查指標,進行正交試驗結果分析。
正交試驗結果見表3。

表3 正交試驗結果
從表3結果可以看出,以乙酸龍腦酯提取率為考查指標,極差結果表明,各因素的作用依次為樣品粒度>提取時間>浸泡時間>溶劑倍數,以樣品粒度因素影響為最大。同時通過直觀分析結果表明,以A3B3C1D3為最佳提取條件。但由于在實際生產過程中,藥材粉碎難度較大,且粉碎后提取液難以過濾,因此樣品的粒度選擇“粉碎”。因此,砂仁藥材的最佳提取條件確定為粉碎15倍,浸泡時間0.5 h,提取時間6.0 h。
4 純化試驗
4.1 提取
稱取砂仁藥材樣品300 g,按正交試驗最佳提取條件進行提取,將樣品破碎,加入15 BV(4 500 mL)純凈水,浸泡0.5 h,加熱回流提取6.0 h。
4.2 濃縮
將提取液趁熱過濾,濾液濃縮至體積約300 mL(生藥質量濃度1.0 g/mL)。
4.3 醇沉
將以上得到的濃縮液分為4份,每份樣品75 mL,相當于原藥材75 g。將其中3個濃縮液中分別加入不同體積的乙醇溶液,使醇沉分別達到60%,70%,80%,攪拌均勻,靜置12 h。其中,1個濃縮液不醇沉,作為對比樣品。
4.4 黃酮提取物
將醇沉液離心后的上清液回收乙醇,濃縮干燥,得到砂仁黃酮提取物。同時將未醇沉的濃縮液直接濃縮干燥,得到砂仁粗提取物,并按試驗方法分別對提取物樣品進行總黃酮含量測定。
砂仁黃酮提取物試驗結果見表4。

表4 砂仁黃酮提取物試驗結果
從表4試驗結果可以看出,醇沉可有效提高提取物中總黃酮的含量,隨著醇沉質量分數的不斷提高,砂仁總黃酮提取物的得率和總黃酮成分的得率越來越低,提取物的含量越來越高。
4.5 多糖提取物
將醇沉液離心后的沉淀直接干燥粉碎,得到砂仁多糖提取物。同時,將未醇沉的濃縮液直接濃縮干燥,得到砂仁粗提取物,并分別按試驗方法對提取物進行總多糖含量測定。
砂仁多糖提取物試驗結果見表5。
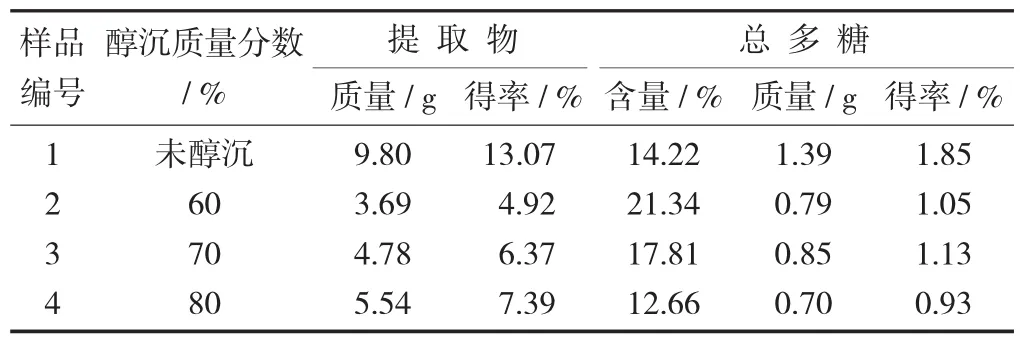
表5 砂仁多糖提取物試驗結果
從表5試驗結果可以看出,隨著醇沉質量分數的不斷提高,砂仁總多糖提取物的得率越來越高,提取物的含量越來越低,但總多糖的得率以70%醇沉質量分數為最高。綜合總黃酮和總多糖提取物的試驗結果分析,砂仁粗提取物的純化條件以醇沉質量分數70%為最適宜。
5 驗證試驗
5.1 試驗條件
稱取砂仁藥材樣品150 g共2份,將樣品破碎,加入15 BV(2 250 mL) 純凈水,浸泡0.5 h,加熱回流提取6.0 h,收集得到砂仁揮發油。將提取液過濾,濃縮至體積約150 mL(生藥質量濃度1.0 g/mL)。濃縮液中分別加入乙醇溶液,使醇沉質量分數分別達到70%,攪拌均勻,靜置。將醇沉液用離心機離心,分別取上清液和沉淀。將醇沉離心后的上清液分別回收乙醇,濃縮干燥,得到砂仁黃酮提取物。將醇沉離心后的沉淀干燥粉碎,得到砂仁多糖提取物。
5.2 提取物得率
計算揮發油、黃酮、多糖3種提取物的得率。各提取物的得率見表6。

表6 各提取物的得率
5.3 有效成分得率
按試驗方法測定揮發油中乙酸龍腦酯的含量、提取物中總黃酮和總多糖的含量,分別計算各有效成分的得率。
各有效成分得率見表7。
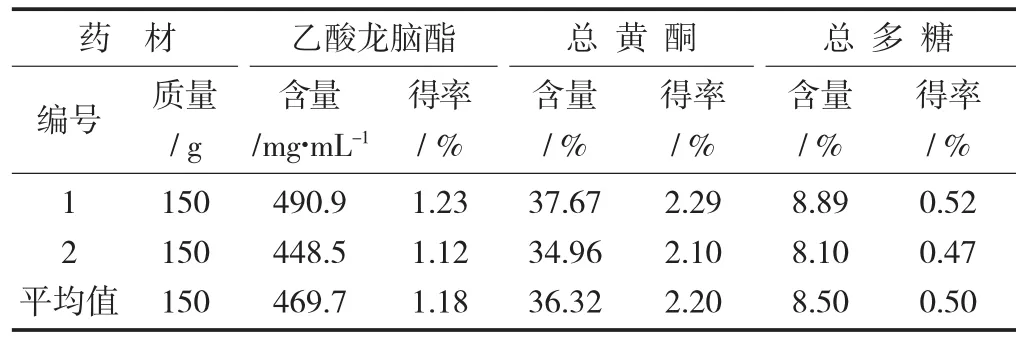
表7 各有效成分得率
6 結論
(1)試驗對砂仁藥材進行加熱提取,通過對水蒸氣冷凝分層收集得到砂仁揮發油,同時對提取液濃縮后醇沉,分別干燥得到砂仁黃酮和多糖提取物,采用一種工藝同時得到砂仁的3種提取物,對砂仁藥材進行了綜合利用,有效降低了生產成本,實現了資源的合理利用,具有顯著的經濟效益和社會效益。
(2)通過提取正交試驗和純化試驗,結合生產的實際情況,砂仁藥材綜合利用最佳試驗條件為取砂仁藥材破碎,加15倍量的水,浸泡0.5 h,提取6.0 h。提取液濃縮至生藥質量濃度1.0 g/mL,醇沉質量分數達到70%。
(3)經過對砂仁藥材綜合利用最佳試驗條件的驗證,每100 g砂仁藥材可得到砂仁揮發油2.5 mL,揮發油中乙酸龍腦酯的含量為469.7 mg/mL。同時可得到砂仁黃酮提取物6.04 g,提取物中總黃酮的含量為36.32%,得到砂仁總多糖提取物5.84 g,提取物中總多糖的含量為8.50%。
猜你喜歡
四川蠶業(2021年2期)2021-03-09 03:15:32
四川蠶業(2021年3期)2021-02-12 02:38:46
中成藥(2018年11期)2018-11-24 02:57:00
中成藥(2017年8期)2017-11-22 03:19:40
中成藥(2017年10期)2017-11-16 00:50:13
中成藥(2017年4期)2017-05-17 06:09:50
哈爾濱醫藥(2016年1期)2017-01-15 13:43:16
天然產物研究與開發(2016年11期)2016-06-15 20:29:17
湖南師范大學自然科學學報(2015年1期)2015-02-27 14:50:04
安徽醫藥(2014年12期)2014-03-20 13:15:15
