動(dòng)物源性食品中青霉素類藥物殘留檢測(cè)
2019-01-08 08:55:00馬燕紅李志剛李瑩瑩郭文萍
食品科學(xué) 2018年24期
關(guān)鍵詞:標(biāo)準(zhǔn)實(shí)驗(yàn)
馬燕紅,王 妍,李志剛,李瑩瑩,郭文萍
(中國(guó)肉類食品綜合研究中心,北京 100068)
近幾十年來(lái)抗生素,尤其是β-內(nèi)酰胺類抗生素,由于其在人類醫(yī)學(xué)和獸藥學(xué)上有效地預(yù)防和治療各種傳染性疾病而備受關(guān)注。青霉素屬于β-內(nèi)酰胺類抗生素,由于其藥效高、價(jià)格低廉、使用方便,已成為臨床上治療細(xì)菌感染最常用的抗生素,在畜牧獸醫(yī)水產(chǎn)養(yǎng)殖領(lǐng)域也得到了廣泛的應(yīng)用。目前主要用于治療奶牛乳腺炎、魚(yú)、蝦細(xì)菌感染和海參的疾病[1]。然而,用藥不規(guī)范對(duì)漁業(yè)生產(chǎn)和生態(tài)環(huán)境都有嚴(yán)重的潛在危害,也會(huì)導(dǎo)致藥物在奶和動(dòng)物、水產(chǎn)品組織中的殘留,使人體產(chǎn)生耐藥性導(dǎo)致免疫力降低,也會(huì)使過(guò)敏個(gè)體存在潛在的風(fēng)險(xiǎn),從而危害人體健康[2]。同時(shí),抗生素殘留也成為國(guó)際貿(mào)易的壁壘[3]。我國(guó)農(nóng)業(yè)部235號(hào)公告規(guī)定芐星青霉素在動(dòng)物源組織中的限量值為50 μg/kg、苯唑西林為300 μg/kg,歐盟指令NO.2377/90/EEC對(duì)青霉素類抗生素在動(dòng)物源性食品中的殘留進(jìn)行了嚴(yán)格限制。所以對(duì)動(dòng)物源性食品中此類抗生素殘留的有效監(jiān)測(cè)已經(jīng)成為保障食品安全的一個(gè)重要環(huán)節(jié)。為滿足殘留檢測(cè)的需要,建立一種動(dòng)物源性食品中青霉素類抗生素殘留的分析方法非常重要。
青霉素類藥物殘留的檢測(cè)方法一直以來(lái)都是科研人員關(guān)注的熱點(diǎn),目前已報(bào)道的方法主要有微生物法[4]、免疫分析法[5-6]、近紅外光譜法[7]、拉曼光譜法[8]、高效液相色譜-紫外法[9-13]、高效液相色譜-熒光法[14]、高效液相色譜-化學(xué)發(fā)光法[15]、液相色譜-質(zhì)譜法[16-19]、電泳法[20]等。其中,微生物法操作快速簡(jiǎn)便、價(jià)格便宜,但專屬性不足,容易產(chǎn)生假陽(yáng)性;免疫分析法由于要選擇配體發(fā)生結(jié)合反應(yīng),線性范圍窄,在檢測(cè)過(guò)程中也易產(chǎn)生假陽(yáng)性;光譜法在定量方面存在缺陷;液相色譜-質(zhì)譜法可避免生物法在準(zhǔn)確性方面的欠缺,靈敏度高、選擇性強(qiáng),但由于儀器價(jià)格昂貴,不能滿足絕大多數(shù)實(shí)驗(yàn)室的需求。高效液相色譜法成為這類藥物殘留應(yīng)用最廣泛的檢測(cè)技術(shù),其準(zhǔn)確性強(qiáng)、靈敏度高,能滿足青霉素類藥物殘留的痕量檢測(cè)需求。
本實(shí)驗(yàn)旨在建立一種動(dòng)物源性食品中青霉素G、苯唑西林、乙氧萘青霉素、雙氯青霉素4 種青霉素類藥物的高效液相色譜檢測(cè)方法。采用乙腈-水提取目標(biāo)化合物,HLB固相萃取柱凈化,高效液相色譜法對(duì)不同基質(zhì)中4 種青霉素類藥物進(jìn)行測(cè)定,并驗(yàn)證了所建方法的選擇性、準(zhǔn)確性、穩(wěn)定性和靈敏性。
1 材料與方法
1.1 材料與試劑
青霉素G鉀鹽(純度99.6%)、苯唑西林(純度91.0%)、乙氧萘青霉素(純度99.0%)、雙氯青霉素(純度97.5%)、水合氨芐青霉素(純度98.5%)德國(guó)Dr. Ehrenstorfer Gmbh公司;乙腈、甲醇(HPLC級(jí)) 美國(guó)Fisher公司;乙酸銨(色譜純) 北京Dikma公司;磷酸氫二鈉、磷酸二氫鉀(均為分析純)國(guó)藥集團(tuán)化學(xué)試劑有限公司。
1.2 儀器與設(shè)備
1260 Infinity高效液相色譜儀(配有二極管陣列檢測(cè)器)、C18色譜柱(150 mm×4.6 mm,5 μm)美國(guó)安捷倫公司;SCR20BA離心機(jī) 日本日立公司;RE52CS-2旋轉(zhuǎn)蒸發(fā)儀 上海亞榮生化儀器廠;Oasis HLB固相萃取柱(500 mg,3 mL)、C18固相萃取柱(500 mg,3 mL) 美國(guó)Waters公司;HGC-36A氮吹儀天津市恒奧科技發(fā)展有限公司。
1.3 方法
1.3.1 標(biāo)準(zhǔn)溶液配制
標(biāo)準(zhǔn)儲(chǔ)備液的配制:準(zhǔn)確稱取青霉素G、苯唑西林、乙氧萘青霉素和雙氯青霉素的標(biāo)準(zhǔn)品各10 mg(精確到0.000 1 g)于10 mL容量瓶中,用水溶解并稀釋至刻度,配制成質(zhì)量濃度為1.00 mg/mL的青霉素G、苯唑西林、雙氯青霉素和乙氧萘青霉素標(biāo)準(zhǔn)貯備液,于4 ℃以下避光保存。
標(biāo)準(zhǔn)工作液的配制:分別量取青霉素G、苯唑西林、雙氯青霉素和乙氧萘青霉素質(zhì)量濃度為1.00 mg/mL標(biāo)準(zhǔn)貯備液各1.00 mL于100 mL容量瓶中,用水溶解并稀釋至刻度,配制成質(zhì)量濃度為10.0 μg/mL的4 種青霉素混合標(biāo)準(zhǔn)工作液,于4 ℃以下避光保存。
內(nèi)標(biāo)儲(chǔ)備液的配制:準(zhǔn)確稱取水合氨芐青霉素標(biāo)準(zhǔn)品10 mg(精確到0.000 1 g)于10.0 mL容量瓶中,用水溶解并稀釋至刻度,配制成質(zhì)量濃度為1.00 mg/mL的水合氨芐青霉素標(biāo)準(zhǔn)貯備液,于4 ℃以下避光保存。
內(nèi)標(biāo)工作液的配制:量取1.00 mL 1.00 mg/mL水合氨芐青霉素標(biāo)準(zhǔn)貯備液于100 mL容量瓶中,用水溶解并稀釋至刻度,配制成質(zhì)量濃度為10.0 μg/mL的水合氨芐青霉素標(biāo)準(zhǔn)工作液。
1.3.2 高效液相色譜條件
C18色譜柱(150 mm×4.6 mm,5 μm);流動(dòng)相A為0.02 mol/L磷酸二氫鉀溶液,流動(dòng)相B為乙腈。梯度洗脫程序:0.00~3.00 min,87% A;3.01~10.0 min,87%~65% A;10.01~15 min,65%~87% A。流速1.0 mL/min;進(jìn)樣量20 μL;柱溫30 ℃;紫外檢測(cè)波長(zhǎng)210 nm。
1.3.3 樣品前處理
提取:準(zhǔn)確稱取切碎均質(zhì)良好的樣品2.00 g于50 mL離心管中,加內(nèi)標(biāo)工作液20 μL,靜置10 min,先加入2 mL水,再加入20 mL乙腈,渦旋混勻30 s,振蕩提取15 min,12 000 r/min離心5 min,將上清液全部轉(zhuǎn)移至旋蒸瓶中,向殘?jiān)性偌铀? mL、乙腈10 mL渦旋混勻,二次提取,將上清液全部轉(zhuǎn)移到旋蒸瓶中。在旋蒸瓶中加入0.1 mL飽和食鹽水,于45 ℃水浴旋蒸至近干,加入3 mL正己烷,渦旋混勻,除去上層正己烷,剩余液體為固相萃取上樣液。
換熱站供熱節(jié)能改造工程一般包括以下內(nèi)容:(1)加裝或更換一次網(wǎng)控制閥、熱量表、遠(yuǎn)傳水表和電表,將換熱站內(nèi)熱、水、電的能源消耗情況進(jìn)行獨(dú)立的統(tǒng)計(jì),建立能耗統(tǒng)計(jì)和分析系統(tǒng),監(jiān)測(cè)換熱站的能耗情況,并制定運(yùn)行調(diào)整方案。(2)二次管網(wǎng)加裝電動(dòng)或手動(dòng)平衡閥,通過(guò)其調(diào)節(jié)性能,提高二次管網(wǎng)的輸配效率,同時(shí)消除管網(wǎng)末端供暖質(zhì)量問(wèn)題,減少因供暖質(zhì)量引起的人為放水。(3)站內(nèi)水泵變頻和PLC控制柜改造,完善站內(nèi)自動(dòng)控制系統(tǒng),增加水泵、閥門的運(yùn)行調(diào)節(jié)方式,加裝或利用站內(nèi)原有溫度、壓力測(cè)點(diǎn)進(jìn)行數(shù)據(jù)采集。實(shí)現(xiàn)一次網(wǎng)閥門多控制方式選擇,同時(shí)滿足上位系統(tǒng)調(diào)整需求。典型換熱站節(jié)能改造示意圖如圖1所示。
凈化:將上樣液轉(zhuǎn)移到HLB固相萃取柱中(使用前用3 mL甲醇、3 mL水和3 mL pH 8.5的磷酸鹽緩沖液活化),再用3 mL磷酸鹽緩沖液洗滌雞心瓶2 次,洗液一并轉(zhuǎn)移到萃取柱中;再用3 mL水淋洗并抽干萃取柱,用2 mL甲醇+2 mL乙腈洗脫并收集于5 mL帶刻度的玻璃瓶中,50 ℃氮吹至小于1 mL,用乙腈定容至1 mL,渦旋混合,過(guò)0.22 μm濾膜,供高效液相色譜分析。對(duì)每一個(gè)待測(cè)樣品平行測(cè)定3 次,取平均值。
1.4 數(shù)據(jù)處理
采用SPSS 16.0統(tǒng)計(jì)軟件進(jìn)行數(shù)據(jù)統(tǒng)計(jì)分析,采用單因素方差分析(ANOVA),數(shù)據(jù)以 ±s表示,P<0.05,表示差異顯著。
2 結(jié)果與分析
2.1 儀器條件的優(yōu)化
由于青霉素類化合物在弱酸性條件下易水解,水解產(chǎn)物中含有巰基、羰基等,易與金屬離子形成絡(luò)合物,國(guó)標(biāo)[21]和文獻(xiàn)[22]因此采用衍生化法測(cè)定此類化合物。但會(huì)用到氯化汞等毒性物質(zhì),操作也較為復(fù)雜。而青霉素類化合物是β-內(nèi)酰胺類,具有苯環(huán)、羰基等發(fā)色基團(tuán),在紫外波長(zhǎng)范圍內(nèi)有吸收峰。故本實(shí)驗(yàn)采用直接進(jìn)樣法檢測(cè)這類化合物。
青霉素類藥物由于具有不穩(wěn)定的四元環(huán),易發(fā)生反應(yīng)。用外標(biāo)法測(cè)定時(shí),回收率較低,故本實(shí)驗(yàn)選取性質(zhì)接近的水合氨芐青霉素作為內(nèi)標(biāo)物用以提高回收率。
本實(shí)驗(yàn)在大量文獻(xiàn)和前期工作的基礎(chǔ)上,對(duì)比乙腈-水、乙腈-0.02 mol/L乙酸銨溶液、乙腈-0.02 mol/L磷酸二氫鉀溶液和乙腈-0.05 mol/L磷酸二氫鉀溶液作為流動(dòng)相,標(biāo)準(zhǔn)物質(zhì)出峰的半峰寬、分離度、保留因子等參數(shù)情況。結(jié)果表明:采用乙腈-水為流動(dòng)相進(jìn)行實(shí)驗(yàn)時(shí),溶劑峰嚴(yán)重拖尾,且化合物不出峰,原因可能是流動(dòng)相中不含鹽,與固定相無(wú)保留;在流動(dòng)相中加入鹽后,化合物與固定相作用,可以正常出峰,但乙腈-0.02 mol/L乙酸銨溶液為流動(dòng)相時(shí),色譜峰的半峰寬大于乙腈-0.02 mol/L磷酸二氫鉀溶液為流動(dòng)相時(shí)的峰寬;加大鹽的濃度,峰形變窄,變尖。但考慮到鹽濃度增大在色譜柱中易結(jié)晶,影響色譜柱壽命,本實(shí)驗(yàn)選取乙腈-0.02 mol/L磷酸二氫鉀溶液作為流動(dòng)相。
以不同體積比的0.02 mol/L磷酸二氫鉀-乙腈溶液作為流動(dòng)相,考察4 種青霉素混合標(biāo)準(zhǔn)溶液的出峰時(shí)間。流動(dòng)相比例為0.02 mol/L磷酸二氫鉀-乙腈(70∶30,V/V)溶液等度洗脫,在12 min內(nèi)可完成分離,如圖1A所示。但內(nèi)標(biāo)峰水合氨芐青霉素在2.6 min左右出峰,青霉素G出峰時(shí)間在3.8 min左右,出峰時(shí)間較早。因動(dòng)物源食品成分復(fù)雜,可能會(huì)對(duì)水合氨芐青霉素、青霉素G的分析產(chǎn)生干擾。因此本實(shí)驗(yàn)選取梯度洗脫,洗脫程序如1.3節(jié)所述,分離效果圖見(jiàn)圖1B。出峰順序依次為水合氨芐青霉素、青霉素G、苯唑西林、乙氧萘青霉素和雙氯青霉素。
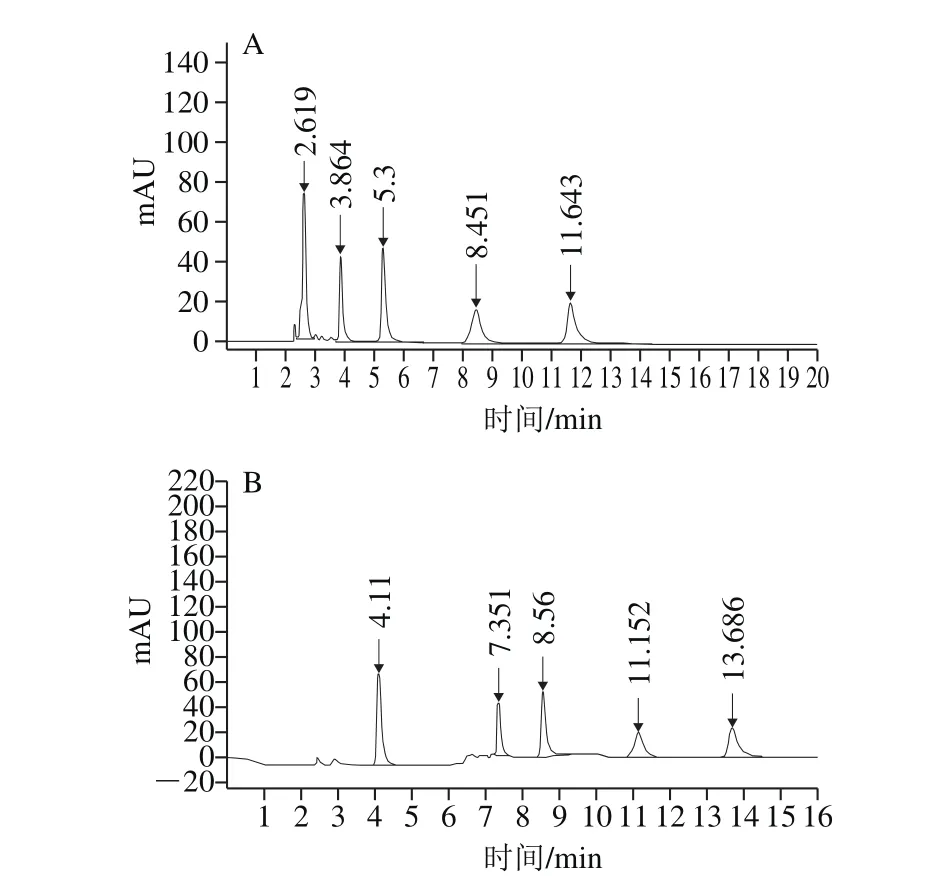
圖1 等度洗脫(A)和梯度洗脫(B)色譜圖Fig. 1 Chromatograms obtained by isocratic elution (A) and gradient elution (B)
2.2 提取條件的選取
2.2.1 提取溶劑的選擇
參照文獻(xiàn)方法,本實(shí)驗(yàn)比較了乙腈[23]、乙腈-水(10∶1,V/V)[24]、乙腈-水(4∶1,V/V)[19]、丙酮-水(1∶1,V/V)[25]作提取溶劑,其他條件如1.3.3節(jié)所述,加標(biāo)量在50 μg/kg時(shí)的回收率,結(jié)果見(jiàn)圖2。用乙腈和乙腈-水(10∶1,V/V)作提取溶劑時(shí),回收率均在90%左右,后者回收率略高,其他2 組回收率較低。可能是由于乙腈提取時(shí)沉淀蛋白完全,易于離心分離,提前加入水能先溶解目標(biāo)化合物。故本實(shí)驗(yàn)選取乙腈-水(10∶1,V/V)作為提取溶劑。減壓蒸餾過(guò)程中加入飽和食鹽水,可以防止乙腈在旋蒸過(guò)程中暴沸[24]。
2.2.2 SPE柱的選擇
由于動(dòng)物源性食品基質(zhì)復(fù)雜,選用SPE凈化小柱獲得較好的凈化效果。青霉素類化合物分子中含有羥基,顯酸性,易溶于甲醇、乙醇、乙腈等有機(jī)溶劑。文獻(xiàn)中廣泛采用的是。本實(shí)驗(yàn)比較了這2 種SPE柱在相同條件下的回收率。加標(biāo)量為50 μg/kg,3 mL甲醇、3 mL水活化,上樣,水淋洗后抽干,最后用2 mL甲醇+2 mL乙腈洗脫,N2吹干后乙腈復(fù)溶。如圖3所示,HLB柱對(duì)目標(biāo)化合物的回收率均高于C18柱,與文獻(xiàn)[24-25]報(bào)道結(jié)果一致。
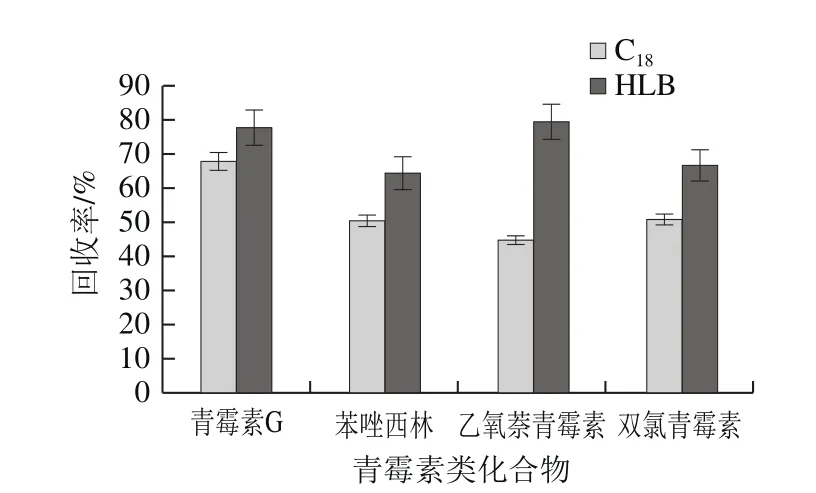
圖3 不同SPE柱的回收率Fig. 3 Effect of different SPEs on recovery of penicillins
2.2.3 SPE洗脫條件的優(yōu)化
不同的淋洗和洗脫溶劑也影響了SPE柱的凈化效果。本實(shí)驗(yàn)在文獻(xiàn)的基礎(chǔ)上,比較不同的淋洗和洗脫溶劑對(duì)回收率的影響,如表1所示。

表1 SPE洗脫條件的優(yōu)化Table 1 Optimization of SPE conditions
從表1可以看出,洗脫液為2 mL甲醇+2 mL乙腈時(shí),4 種化合物的回收率明顯提高。當(dāng)在活化和淋洗劑中加入堿性磷酸緩沖鹽時(shí),回收率達(dá)到了90%左右。可能是堿性磷酸鹽緩沖液防止待測(cè)物降解[12]。最終確定SPE萃取小柱的最佳條件為:選取HLB萃取柱,采用方案4的方式凈化。
2.3 方法的驗(yàn)證
2.3.1 標(biāo)準(zhǔn)曲線、檢出限和定量限結(jié)果
配制質(zhì)量濃度分別為0.01、0.05、0.10、0.50、1.00、2.00 μg/mL 6 個(gè)水平4 種青霉素的標(biāo)準(zhǔn)混合溶液,含內(nèi)標(biāo)0.20 μg/mL,待高效液相色譜分析。每個(gè)溶液分別進(jìn)樣3 次,以目標(biāo)化合物與內(nèi)標(biāo)質(zhì)量濃度之比對(duì)其峰面積之比繪制4 種青霉素標(biāo)準(zhǔn)曲線,其中X為青霉素與內(nèi)標(biāo)物的質(zhì)量濃度比,Y為二者峰面積之比。從表2可以看出,各目標(biāo)化合物標(biāo)準(zhǔn)曲線的相關(guān)系數(shù)均大于0.999,顯示了良好的相關(guān)性。檢出限和定量限采用空白樣品中添加青霉素類化合物的方法,分別以信噪比不小于3和信噪比不小于10計(jì)算。從表2可以看出,各青霉素的檢出限為3 μg/kg,定量限為10 μg/kg,可以滿足青霉素類化合物的定性及定量測(cè)定要求。

表2 4 種青霉素的標(biāo)準(zhǔn)曲線及其相關(guān)系數(shù)、檢出限和定量限Table 2 Linear ranges, calibration curves, LODs and LOQs for four penicillins
2.3.2 精密度測(cè)定結(jié)果
在空白豬肉樣品中進(jìn)行精密度實(shí)驗(yàn),樣品添加不同含量的標(biāo)準(zhǔn)溶液后,渦旋混勻,于室溫放置10 min,使待測(cè)成分與樣品機(jī)體成分互相作用達(dá)到平衡,按1.3.2節(jié)和1.3.3節(jié)所述方法,做6 次重復(fù)實(shí)驗(yàn),測(cè)定結(jié)果的相對(duì)標(biāo)準(zhǔn)偏差在10%以內(nèi)。
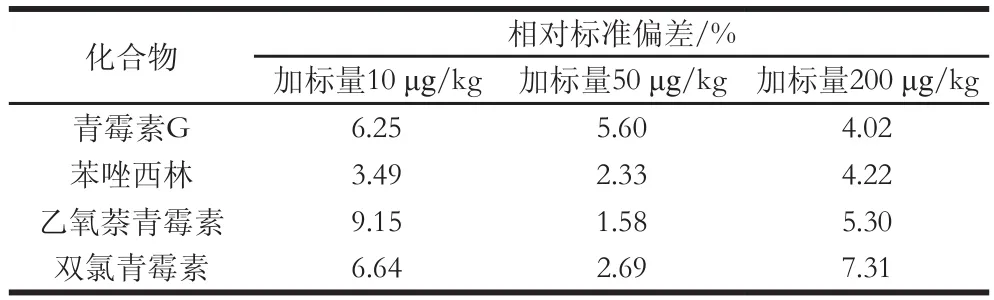
表3 精密度實(shí)驗(yàn)(n=6)Table 3 Results of precision test (n=6)
2.3.3 回收率測(cè)定結(jié)果
在空白樣品中分別加入低、中、高3 個(gè)質(zhì)量濃度的標(biāo)準(zhǔn)品進(jìn)行回收率計(jì)算,選擇分別加入10、50、200 μg/kg 4 種青霉素混合標(biāo)準(zhǔn)工作液,利用上述確定的檢測(cè)方法,每個(gè)加標(biāo)量做3 次平行實(shí)驗(yàn)。由表4可知,在不同的基質(zhì)中,3 個(gè)水平的回收率在69.9%~100.3%范圍內(nèi)。從精密度和回收率的結(jié)果可以得出,該方法有較高的準(zhǔn)確性和可靠性。
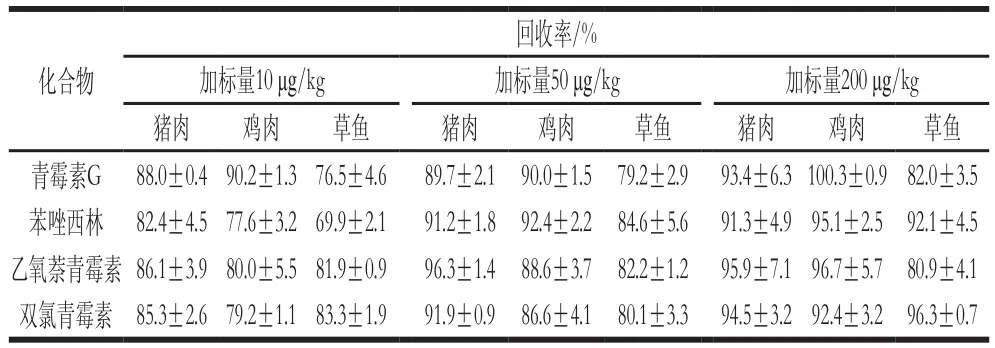
表4 4 種青霉素低、中、高3 個(gè)水平的回收率Table 4 Recovery rates for four penicillins at three different spiked levels
2.3.4 實(shí)際樣品驗(yàn)證
從市場(chǎng)中選取5 種基質(zhì)進(jìn)行方法驗(yàn)證,分別為豬肉、牛肉、羊肉、雞肉、草魚(yú),目標(biāo)化合物出峰處均無(wú)干擾、未檢測(cè)出4 種青霉素類藥物殘留。
3 結(jié) 論
本實(shí)驗(yàn)以水合氨芐青霉素作為內(nèi)標(biāo),建立一種高效液相色譜法檢測(cè)動(dòng)物源性食品中的青霉素G鉀鹽、苯唑西林、乙氧萘青霉素、雙氯青霉素4 種青霉素類藥物殘留量的方法。采用直接進(jìn)樣,無(wú)需衍生化,內(nèi)標(biāo)法定量提高了回收率。該方法前處理簡(jiǎn)單、快速、準(zhǔn)確、靈敏,可以滿足實(shí)際檢測(cè)分析的需要。
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學(xué)生數(shù)理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學(xué))(2019年6期)2019-10-10 01:01:50
當(dāng)代陜西(2019年8期)2019-05-09 02:22:48
動(dòng)漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(shù)(2018年4期)2018-05-09 07:07:52
發(fā)明與創(chuàng)新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
專用汽車(2016年4期)2016-03-01 04:13:43
