ATM缺失阻礙骨骼肌蛋白質合成的研究進展
2019-01-17 03:01:42馮祎中
浙江體育科學 2019年1期
關鍵詞:胰島素
金 晶,馮 燕,馮祎中
(1.浙江農林大學 體育軍訓部,浙江 杭州 311300;2.華東師范大學 體育與健康學院,上海 200241)
惡性體質(cachexia)是一種逐漸被認知的慢性病發生發展的疾病綜合體,包括慢性心力衰竭、慢性腎病、慢性阻塞性肺部、骨骼肌衰減征等[1]。惡性體質不但引起財政和醫療的巨大負擔,有較多的報告顯示惡性體質與身體質量指數(body mass index,BMI)和骨骼肌丟失有高度相關性[2,3],且增加個人致殘率甚至導致死亡[1]。Arthur[4]報告美國超過160 000惡性體質的住院案例,惡性體質人群住院時間中值為6天,平均消費超過10 000美元/人次,而非惡性體質住院者中值為3天,平均消費為4 000美元/人次。古希臘名醫希波克拉底(公元前460-377年)描述心力衰竭病人寫到:肉體被分解并開始化作水,腹部充水、腿腳腫脹,肩膀、鎖骨、胸部和大腿被融化,這種病是致命的[5]。2009年德國諾貝爾文學獎獲得者赫塔米勒同樣冷淡地提及:當肉體逐漸從身體上消失,馱著骨架已成負擔,驅使你埋進土地[6]。
毛細血管擴張共濟失調(ataxia telangiectasia,AT)也是惡性體質的一種,案例研究報告AT病人的身材矮小生長停滯[7]、骨骼肌力下降[8],毛細血管擴張共濟失調突變(ataxia telangiectasia mutated,ATM)與骨骼肌蛋白質合成有關[9]。盡管文獻對ATM與mTOR通路的氧化應激[10]和DNA損傷[11]有所描述,但在ATM與蛋白質合成的分子機制描述并不全面,如Ching JK[9]只研究ATM對IGF-1/PI3K/Akt通路的影響,Burnett PE[12]只針對研究ATM對mTOR信號通路下游信號分子的影響,相關研究都提示ATM通過干預蛋白質合成呈現個體身材矮小、骨骼肌丟失等現象。本文擬通過對AT疾病和骨骼肌相關表型回顧,綜述ATM與相關分子的調控關系,系統描繪ATM阻礙骨骼肌蛋白質合成的可能途徑。
1 AT疾病和ATM
1.1 AT的癥狀
1994年統計美國和英國地區的發病率評估在1/40 000—1/100 000,AT雜合型女性擁有很高比例的乳腺癌風險,攜帶者的頻率為0.5%~1.0%[13],在美國高加索人群中比例高達2%[14],現階段還沒有行之有效的治療手段。針對美國地區出生兒童進行統計,報告發病率在1/40 000~1/300 000,并且疾病表型發生于幼兒時期,尤其在2~5歲已開始[15]。AT疾病是進程性的,隨著年齡增長淋巴瘤和神經惡化進而增加死亡率和致殘率,平均預期壽命在25歲[16,17]。
在AT基因被定以前,所有的文獻都認為AT病人有極高的小腦共濟失調和眼部及皮膚毛細血管擴張的發生率(100%的案例)[18,19]。然而,在AT基因被定義之后,有很多文獻發現AT基因的病例沒有發生小腦共濟失調或眼部的毛細血管擴張:Trimis[20]等報告6歲女孩遺傳學上被確認為AT病人,但是沒有發生神經系統的病變;Alterman[21]等研究兩兄妹發現雖然有嚴重的細胞表型,但只存在輕微的神經臨床表型;moin[22]等評定臨床和實驗室的104名AT病人,發現所有病人都存在小腦的共濟失調,而眼球和眼部皮膚的毛細血管分別發生87和73人。
1.2 ATM的發現與定義
毛細血管擴張共濟失調突變(ataxia telangiectasia mutated,ATM)是常染色體上隱性基因不穩定發生突變形成以小腦共濟失調、免疫缺失、癌癥和呼吸系統易感染等特征的綜合癥[23],稱為“A-T”(ataxia telangiectasia,毛細血管擴張共濟失調)。AT還叫作路易斯-巴爾二氏綜合癥,也經常用艾琳娜博德和羅伯特塞奇威克,第一次被描述為家族性的退行性小腦失調癥狀[24]。ATM基因位點最早是由Gatti[25]等通過數理分析共分離統計定位于第11號染色體的q22.3-23.1,在隨后的七年,許多研究所和科研工作者不斷完善,最終由Savitsky[26]團隊將其定義完成,它由66個外顯子(4個非編碼和62個編碼)跨越150kb的DNA組成。ATM是~350kDa的高分子蛋白激酶,屬于磷脂酰肌醇-3激酶相關蛋白激酶(phosphoinositidyl 3-kinase-related protein kinase,PIKK)家族成員[27],認為它在調控細胞循環和DNA損傷方面有重要作用[28,29]。
2 AT與生長、骨骼肌和運動能力
2.1 AT與身高、體重
絕大多數AT患者都發現有生長停滯和體重下降等特征[7,8,30-33]。研究報道AT病人的身高、體重以及身體質量指數(body mass index,BMI)均顯著低于對照組[32,34]。Ehlayel[35]和Pommerening[8]等分別報告13名和25名AT病人與對照組比較,發現身高存在極其顯著性差異。澳大利亞地區的13名AT病人(年齡4~23歲,其中女性9人)結果顯示77%病人體型偏小(z score<-1),且54%的病人體重不足[36]。Pommerening[8]等研究發現AT病人的體重和BMI極其顯著低于對照組,Schubert R[33]將19名AT患者(男性11人和女性8人,年齡為4~24歲)按年齡分為三組,A組≤12歲、B組12~18歲和C組>18歲。結果顯示A組的BMI為15.3 kg/m2,B組的病人顯示體重和生長發育困難引起BMI下降到正常范圍內的3%之前,C組出現進一步下降的惡性變化,BMI盡然為12.7 kg/m2。與此結果相一致的研究還有英國地區70名AT病人的統計研究,54/70的病人出現極其偏瘦[30];Kieslich M[31]團隊對11名病人(8~26歲)進行體重和BMI的分析結果均顯示是顯著低于對照組(P<0.05);Schubert R[33]對19名AT病人盡管給予充足的營養條件,BMI仍然顯著下降。
另外,AT的動物模型通過敲除ATM基因即ATM-/-小鼠個體,同樣也出現生長延緩或停滯現象[37,38]。James KC團隊發現ATM敲除小鼠純合子具有小體型,而雜合子ATM+/-與野生型小鼠體重沒有差異性,而ATM-/-小鼠的體重是野生型的70%,與AT病人體重下降結論相一致[9]。
2.2 AT與骨骼肌及運動能力
骨骼肌是人體構造的重要組成部分,是氨基酸合成蛋白質的儲水庫[39]。厭食、脫水、惡性體質和骨骼肌衰減征等引起骨骼肌丟失最常見的原因,造成肌肉進程性的下降病理生理學的原因是復雜和多因素的,比如久坐不運動、疾病、衰老和營養狀況等都有因果聯系[40]。近期報告提示ATM與骨骼肌丟失存在很大的相關性,發生骨骼肌的丟失后進而直接或間接影響個體的運動能力[7,8,34]。
2.2.1 AT與骨骼肌。研究報告許多疾病所構成的惡性體質(cachexia)都會造成骨骼肌質量的大量丟失,進而呈現個體運動能力受限和功能狀態的低下[1]。例如心力衰竭病人的臨床表現為心肌衰竭,限制其運動能力,骨骼肌質量和力量下降從而影響其運動能力[2];Martin L[3]等研究證明所有癌癥病人均出現體重下降現象,肌肉指數下降和肌肉衰減的癥狀;AT患病個體無論是個案研究還是群體調查都發現體重和骨骼肌力下降及肌肉衰減等癥狀[7,8]。
Da silva R[41]在研究AT病人時發現有很高比例患者出現降低瘦體重(Fat Free-mass,FFM)現象,Pommerening H[8]同樣也發現FFM在AT人群中含量顯著低于對照組,并且認為AT病人的脂類和脂肪組織沒有發生變化,也就是說體重下降主要是減少骨骼肌質量。Heil JA報告4名來自兩個家庭的成年AT病人,患有脊椎肌肉萎縮癥并伴有股四頭肌的萎縮;Dunn HG[42]針對加拿大AT病人進行尸檢,發現骨骼肌出現輕微的萎縮現象。利用液壓測力計測定AT病人的力量顯著低于對照組[8],Felix E[32]發現病人的初始呼吸肌的力量和肺活量顯著低于對照組,經過24周的呼吸肌訓練,有效的加強肺活量和呼吸肌力量,極大改善AT病人的生活質量。
2.2.2 AT與運動能力。英國70人案例研究發現,43/68大部分患者報告3歲之前發生運動失調癥狀[30],另一項最新的個案研究顯示病人從34周齡出生、6月坐、11月站立、14月行走都是正常的,而從兩歲開始行走能力發生改變,需要外力的支撐、身體開始呈現不穩定狀態及頻繁摔倒等現象出現[7],與gatti RA[43]報告運動失調發生在2~3歲吻合。Teive HA[14]等在研究AT的綜述中表述,5~10歲出現更為嚴重的神經性病變導致運動能力的缺失、徐動癥的發生。Felipe RB[7]等報告的個案在6歲時出現進階式的運動失調和顫抖,7歲時完全依靠外界控制站立,四肢末梢開始顫抖,只能依靠某一姿勢控制身體;較早的文獻也報告AT病人手臂出現顫抖現象而且精準度下降,行走困難有搖晃狀態,站立和坐姿都顯示發軀干彎曲等現象[42]。Woods CG[30]統計研究認為病人在8歲失去書寫能力,平均10歲左右失去行走能力。年齡對于AT病人的行走能力有很大相關性,行走組(gait preserved)和輪椅組(wheelchair-bound)之間年齡存在顯著差異;AT病人的去脂體重和體細胞質量極其顯著低于對照組;維生素D的含量AT病人顯著低于對照組,有趣的發現所有缺乏維生素D的病人都是大于12歲并且失去行走能力全部為輪椅組的[8]。
3 抗阻運動誘導骨骼肌肥大及其合成機制
3.1 抗阻運動誘導骨骼肌肥大及IGF-1的重新分配
抗阻運動是肌肉受到劇烈收縮進而刺激骨骼肌肥大的有效方式[44,45]。Luciano TF[44]構建3種不同的抗阻模型對比各方案中大鼠骨骼肌橫截面積、質量和蛋白質合成等,結果顯示所有抗阻模型經過訓練后骨骼肌的體積得到顯著提升;Tang JE[46]招募普通青年男子通過12周的抗阻訓練后,骨骼肌活檢前后比較,發現骨骼肌的橫截面積顯著增加;Salvadego D[47]則通過長期從事健美的運動員與對照組進行對比,結果顯示股四頭肌的橫截面積和體積顯著大于對照組(P<0.05);Ogasawara[45]等采用動物模型通過電刺激模擬抗阻運動,研究發現腓腸肌的凈含量和相對體重比值,單次急性刺激后均沒有變化,而通過12次和18次訓練后分別增加8.6%和10.7%。
普遍認為抗阻運動是通過IGF-1/PI3K/Akt通路激活mTOR,繼而促進骨骼肌的蛋白質合成[48]。Arnarson A[49]觀察全身骨骼肌中胰島素樣生長因子(insulin-like growth factor-1,IGF-1)的分布情況,發現IGF-1經過12周抗阻運動干預后有很強的變化,下調血清中總IGF-1的含量。血清IGF1經過抗阻運動后實驗報告下調或不變:Mangine[50]對比高強度與中等強度抗阻運動,研究結果顯示高強度組的血清IGF-1顯著下調;另外的研究認為抗阻運動后未造成血清IGF-1的改變[51,52]。相對于血清的變化,骨骼肌中的IGF-1顯著上調,Kido[51]實驗通過急性抗阻運動后,測試1h和3h后骨骼肌的IGF-1發現顯著上調;相似的另一項研究檢測骨骼肌IGF-1濃度在1h和6h后上調[52]。抗阻運動干預實驗,總體血清IGF-1始終保持不變或下調,而骨骼肌中IGF-1上調,推測抗阻運動誘導骨骼肌中IGF-1在全身的重新分配,特別是骨骼肌中高表達的IGF-1參與骨骼肌蛋白質合成過程[49,50]。
3.2 骨骼肌蛋白質合成經典機制
運動誘導骨骼肌生理性適應肥大機制涉及到多種信號傳導途徑,其中雷帕霉素靶體蛋白(mammalian target of rapamycin,mTOR)信號傳導通路目前備受關注[53]。mTOR是非典型絲氨酸/蘇氨酸蛋白激酶,在進化上高度保守,其分子量大小是289kDa,是磷脂酰肌醇激酶相關激酶PIKK蛋白家族成員[54]。mTOR具有廣泛的生物學功能,可整合營養、能量及生長因子等多種細胞外信號,參與基因轉錄、蛋白質翻譯、核糖體合成等過程,在調控蛋白質合成、細胞增殖、凋亡、自噬等細胞生長過程中發揮及其重要的作用[54]。
大量的文獻證明mTOR在調控蛋白質合成和骨骼肌細胞增長/肥大過程中起到關鍵作用[53,55,56]。以mTOR為關鍵節點本文將mTOR上游信號通路(IGF-1/PI3K/Akt通路和TSC1/2通路)和下游信號通路(4E-BP1/eIF4E通路和S6K1通路)分別進行詳述。
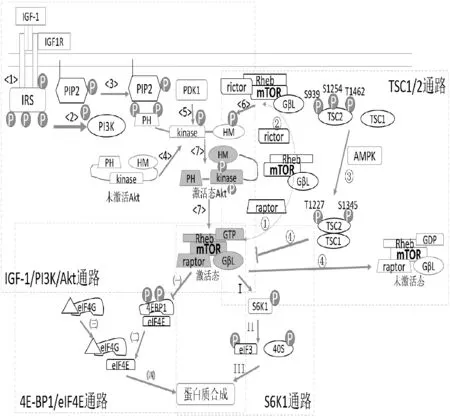
圖1 導致骨骼肌細胞肥大的主要胞內信號傳導途徑(綜合參考文獻[57-59]中的各圖重新繪制)
3.2.1 IGF-1/PI3K/Akt通路。在IGF-1介導的通路中發生信號級聯放大,這種放大反應起初發生在細胞膜上,由IGF-1與IGF-1受體(insulin-like growth factor-1 receptor,IGF-1R)相結合,可引起IGF1R的磷酸化。IGF1R自身磷酸化后,激活細胞內酪氨酸激酶進而促使下一靶點蛋白胰島素受體底物(IRS)的磷酸化[48,60](圖1,途徑<1>)。該反應引起IRS的大量募集,IRS是IGF1R和PI3K(磷脂酰肌醇-3-激酶)之間的銜接蛋白,其磷酸化能進一步激活PI3K[9](圖1,途徑<2>)。被激活的PI3K可以催化位于細胞膜磷脂雙分子層內層上的PI(4,5)P2肌醇環3位上繼續添加一個磷酸基團形成PI(3,4,5)P3(圖1,途徑<3>)。骨骼肌細胞膜的內膜上一旦有PIP3的形成,呈未激活態的Akt就會大量被募集到胞漿內膜上,并使其被激活[61]。Akt又稱為PKB(蛋白激酶B)是一類絲氨酸/蘇氨酸蛋白激酶,完整的蛋白質由三大部分所構成:氨基端的pleckstrin homology(簡稱PH)區、中間的激酶(kinase)區和羧基端的疏水作用(簡稱HM)區。在未激活態/原始態的三級構象中,Akt的PH區和HM區距離相對較近,當其PH區結合到膜內層的PIP3上后,整個蛋白質分子呈現平鋪拉伸構象,促使激酶區與HM區得到充分暴露(圖1,途徑<4>)。隨后,Akt上兩個氨基酸位點:激酶區上的蘇氨酸308(圖1,途徑<5>)和羧基端的HM區上的絲氨酸473均被磷酸化(圖1,途徑<6>),從而使Akt完全被激活,最終通過IGF-1/PI3K/Akt通路使mTOR進行磷酸化促其活性被激活[48](圖1,途徑<7>)。
3.2.2 TSC1/2通路。在哺乳動物細胞中mTOR可以分為兩種復合物mTORC1和mTORC2。兩種復合物之間的區別在于組成的亞結構不同,mTORC1包含raptor(regulatory associated protein of mTOR)、GβL(G-proteinβ-subunit-like protein)、Rheb(ras homolog enriched in brain)和mTOR(圖1,途徑①),而mTORC2含有rictor(rapamycin-insensitive companion of mTOR)和其他組成(圖1,途徑②)。兩者都有特定而且唯一的下游靶蛋白,如mTORC1只對4E-BP1和S6K有磷酸化作用,而mTORC2只磷酸化蛋白激酶Cα和Akt[62]。Raptor結合于4E-BP1和S6K的區域作為mTOR信號基序,募集mTORC1復合物,通過mTOR進行磷酸化作用。因此可以認為raptor是緊密連接mTOR與其底物的銜接分子[57]。Rheb是GTP的激酶(GTPase),其結合GDP或GTP決定mTOR是否被激活,與GTP結合時對mTOR進行激活,與之相反,形成Rheb-GDP復合物時則抑制mTOR活性。mTOR復合體與GDP/GTP結合取決于GTPase 激活蛋白(GAP)tuberin(又稱為TSC2)和另一種復合蛋白hamartin(TSC1)[63](圖1,途徑③)。研究發現,在AMPK的參與下TSC1和TSC2形成復合體TSC1/2可調控Rheb與GTP結合向與GDP結合方向轉化,促使Rheb與GDP結合從而使mTOR活性抑制(圖1,途徑④);而磷酸化的Akt可以抑制TSC1/2復合體的活性,進而向Rheb與GTP結合的方向轉變從而激活mTOR,增強其信號通路[57]。
3.2.3 4EBP1/eIF4E通路。mTOR的促合成通路依賴于下游信號4EBP1磷酸化后調節eIF4E的活性,用來提高翻譯效率,進而促進翻譯的起始過程和延伸過程,增加骨骼肌蛋白合成[64](圖2)。
eIF4E是翻譯啟始因子,也是mTOR下游通路的重要蛋白,mRNA翻譯的啟始階段是整個翻譯過程的關鍵部分。活化的mTOR蛋白中含有的raptor結構可以直接/間接的方式結合TOR信號基序的4EBP1的C端,使其4EBP1磷酸化[65]。這個過程中eIF4E首先在5’mRNA末端與m7GTP的cap結構結合,eIF4E還與其他多種亞結構結合(如eIF4A和eIF4G)形成復合體,進而與40S核糖體結合發揮其作用[65]。4EBP1和eIF4G均能與eIF4E相結合而且反應發生是在相同部位,兩者競爭同一底物即eIF4E是它們共同的結合靶點。mTOR至少可以磷酸化4EBP1的Thr37和Thr46兩個位點[57],4EBP1通過磷酸化水平的高低來調控與eIF4E的結合情況,當4EBP1不被mTOR磷酸化時,低磷酸化狀態的4EBP1與eIF4E具有較高的親和力,4EBP1就與eIF4E結合;而當4EBP1被mTOR磷酸化后抑制它們結合(圖1,途徑㈠),處于較高磷酸化狀態時降低了相互之間的親和力則釋放出eIF4E[59](圖1,途徑㈡),eIF4G則搶占作用靶點與eIF4E結合(圖1,途徑㈢)。eIF4E和eIF4G結合并與其他蛋白共同形成復合體和40S共同編碼mRNA,最終促進骨骼肌中蛋白質的合成[58](圖1,途徑㈣)。
3.2.4 S6K1通路。mTOR的促合成效應除了4EBP1/eIF4E通路以外還需要通過S6K1磷酸化來實現。mTOR的下游信號S6K1磷酸化后,能促使5’TOPmRNA翻譯,提高mRNA的翻譯能力[66](圖2)。
整個啟動復合物結構非常復雜,主要由3個核心部分組成(圖2):①eIF4E,結合于m7GTP(7-甲基-GTP)5’覆蓋的mRNA,促進復合物與核糖體一起裝配;②分子腳手架eIF4G,在啟動子和40S核糖體亞結構之上的元件;③ATP依賴的RNA解旋酶eIF4A[67]。4E-BP1和S6K1是mTOR共同的磷酸化靶點,通過對下游的調控結合完成蛋白質合成。
通常情況下S6K1是附著在eIF3復合物上的,與mTOR/raptor共同結合于復合物上。在生長因子、營養物質等條件的作用下,mTOR激活后會使得S6K1上的Thr389磷酸化(圖1,途徑Ⅰ),磷酸化的S6K1隨即就與eIF3復合物脫離。PDK1雖然不是復合物的一部分,但是它能與脫離的Thr389磷酸化的S6K1相互反應,催化作用發生下再次磷酸化S6K1的Thr229[59]。激活態的S6K1進一步磷酸化40S和eIF4B,一方面S6K1直接磷酸化40S核糖體蛋白S6[65];另一方面磷酸化后的eIF4B被募集到eIF3(圖1,途徑II),最終形成復合體發揮編碼mRNA的作用[59](圖1,途徑III)。
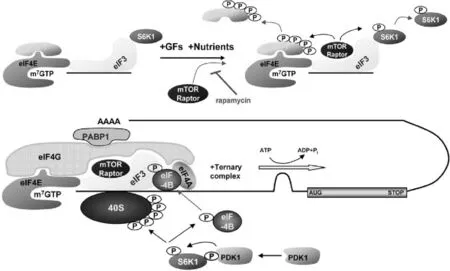
圖2 mTOR下游特色效應器(4E-BP1和S6K1)的反應機制[59]
4 ATM影響蛋白質合成的相關分子機制
4.1 AT病人與IGF-1
在研究AT病人的血清指標發現大部分個體的胰島素樣生長因子-1(Insulin-like growth factor-1,IGF-1)含量普遍偏低,認為IGF-1及其相關蛋白可能是影響生長狀況和體重下降的原因[31, 33]。Schubert R[33]測得9/16病人的IGF-1含量低于同齡對照的前3%,而且13/16病人的IGFBP-3(它是IGF-1主要結合蛋白)濃度顯著較低。Voss S[68]對24名患者進行IGF-1的測定,發現其中10位(41.7%)的濃度低于同齡前3%,Kieslich M[31]也報告6名病人的含量低于前3%。Pommerening H[8]對激素指標進行測定,結果顯示IGF-1、皮質醇和脫氫表雄酮顯著低于對照組。從AT病人獲得的成纖維細胞進行培養,IGF-1-sCLU(secretory clusterin)蛋白水平顯著低于對照組,推測是由于IGF-1含量偏低造成這種現象[69]。
4.2 ATM與胰島素/IGF-1刺激下的Akt磷酸化
Akt和ATM基因敲除模型小鼠顯示如生長停滯、不育癥、免疫系統缺陷和胰島素抵抗等相似的表型[37,70]。一方面,磷酸化的Akt通過IGF-1/PI3K/Akt通路使mTOR進行磷酸化促其活性被激活[48],另一方面,磷酸化的Akt可以抑制TSC1/2復合體的活性,進而促進Rheb與GTP結合的方向轉變從而激活mTOR[57],因此Akt的激活對于骨骼肌蛋白質合成至關重要。
ATM蛋白含量與細胞中Akt磷酸化水平在正常情況下并沒有相關性,當受到胰島素/IGF-1等條件的刺激下ATM的缺失直接影響Akt磷酸化[9,71,72]。Ching JK[9]利用10nM的IGF-1對比目魚肌細胞和C2C12肌管進行20min的刺激,發現ATM KD的C2C12肌管顯著下調Akt的S473/T308磷酸化水平;對比于ATM+/+小鼠的比目魚肌細胞,ATM+/-的Akt S473/T308磷酸化水平發生明顯的阻礙作用,進一步檢測p-mTOR蛋白含量也觀測到其顯著下調,而總體mTOR沒有變化;Ching JK[72]的另一項研究利用2mU/ml的胰島素30min的刺激下,比目魚肌中(ATM+/+和ATM-/-)兩種基因型之間Akt的磷酸化沒有差異性,而脛骨前肌的純合型阻礙Akt兩位點S473/T308磷酸化相較于野生型(P<0.05)。同樣的Jeong[71]利用C2C12 ATM shRNA在100nM胰島素的刺激下,直接阻礙Akt的磷酸化;Hresko RC[48]和Viniegra JG[73]對Cos細胞的ATM+/+和ATM KD進行分析,發現Akt S473的磷酸化在野生型中表達明顯升高,AT病人(GM08931)和KO小鼠獲得的細胞系在胰島素的刺激下抑制Akt的S473磷酸化作用[73];小鼠的胚胎成纖維細胞模型ATM+/+和ATM-/-,也證明了ATM的缺失對Akt S473的磷酸化作用[74]。
KU55933是ATM的抑制劑,阻礙胰島素刺激的Akt的磷酸化。Halaby MJ利用KU55933抑制小鼠L6肌細胞,抑制Akt的磷酸化[74];Jeong利用1μM的KU55933分別對L6肌管、C2C12、比目魚肌和RD(分化后的橫紋肌肉瘤)細胞進行干預,發現C2C12和RD細胞中抑制ATM使得Akt的S473磷酸化有明顯的下調作用,L6肌管細胞在1μM KU55933+100nm/25nm的胰島素都沒有對Akt磷酸化有影響[71]。過量表達的ATM促進野生型誘導S473的磷酸化,但在ATM KD型中沒有變化,Akt的T308磷酸化也沒有差異性;另一種方式通過siRNA技術外源性感染細胞抑制ATM的表達,結果顯示顯著下調S473的磷酸化[73]。綜上所述,ATM被抑制/缺失則下調Akt的磷酸化,而當過量表達時上調其磷酸化水平,因此認為ATM在胰島素/IGF-1的刺激下正調控Akt的磷酸化水平,特別是S473磷酸化水平。
4.3 ATM與Akt的上游信號通路
4.3.1 ATM與IGF-1R。IGF-1R是IGF-1的受體,是酪氨酸激酶的跨膜受體幾乎所有組織中都表達,調控細胞的生長、分化、轉化和凋亡有重要作用[75]。IGF-1R基因缺失也會造成體重下降,IGF-1R-/-小鼠的體量與野生型比較下降45%,出生后的模型鼠多個器官異常并導致死亡,另外IGF-1R-/-的小鼠胚胎時期普遍發生器官和骨骼肌的發育不良[76];另外兩項細胞實驗都認為ATM對IGF-1/IGF-1R有影響,Luo X[69]從AT病人獲得的成纖維細胞進行細胞培養,發現突變細胞的IGF-1-sCLU(secretory clusterin)水平顯著低于對照組,認為ATM缺失誘導IGF-1含量偏低造成這種現象,Goetz EM[77]將正常細胞暴露于DNA損害元件時發現提升IGF-1的表達量,進而激活IGF-1R通路和sCLU,然而ATM缺失的細胞不能誘導IGF-1R的表達。
Peretz S[78]在研究AT細胞時發現與雜合型/野生型相比,IGF-1R處于較低水平(P<0.05)。為了探尋ATM與IGF-1R的直接關系,轉染完整ATM全片段的cDNA到AT突變細胞中[79],8個細胞GM5849通過實驗技術Western blot驗證cDNA全部成功轉染,證明有完整表達的ATM cDNA能提高IGF-1R的表達量[78]。緊接著,在AT細胞中發現IGF-1R啟動子活性下降,為了證明轉染ATM cDNA的細胞中IGF-1R啟動子區域活性也增強,將該區域用熒光素酶受體轉染[80],結果顯示轉染后的ATM cDNA結合細胞中啟動子的活性是ATM突變細胞的4倍;另一個實驗利用ATM突變(GM3487)和父母雜合型(GM3489)獲得的成纖維細胞進行啟動子區域的熒光素酶轉染,結果也證實了ATM+/-的啟動子活性表達量顯著高于ATM-/-[78]。Peretz S[78]利用轉載體的轉染到野生型成纖維細胞系中,與ATM蛋白的亮氨酸拉鏈結構相結合,抑制ATM蛋白的產生,結果證明ATM受到抑制后下調IGF-1R的表達量。
4.3.2 ATM與IRS-1和PI3K。IRS-1是IGF-1R的靶點,可作為IGF-1R和PI3K的銜接蛋白。從同一樣本的不同位點中的p-IRS Y612和總IRS分別進行檢測,結果顯示C2C12細胞的野生型和雜合型在添加IGF-1刺激情況下p-IRS和總IRS-1都有顯著性差異提高,胰島素刺激下P-IRS-1/IRS-1的比值在雜合型中表達偏低,未達到統計意義[9]。胰島素信號通路會被氨基末端激酶(Jun N-terminal kinase,JNK)誘導的IRS-1 S307的磷酸化所阻斷,在ATM-/-動脈細胞發現JNK/總JNK和IRS-1 S307磷酸化/總IRS-1顯著高于ATM+/-和ATM+/+,ATM缺失小鼠的脂肪組織、骨骼肌細胞以及肝臟都存在高活性的JNK,激活IRS-1 S307磷酸化阻礙胰島素信號通路[81]。
Ching JK[9]利用激酶反應試劑盒檢測比目魚肌中野生型和雜合型的PI3K活性,結果顯示IGF-1刺激下的PI3K活性在ATM+/-中偏低(P=0.053)。Viniegra JG[73]研究ATM對Cos細胞胰島素刺激的Akt S473的磷酸化有直接作用,進一步探究是否通過抑制PI3K能夠阻礙Akt的磷酸化,與預期的相一致,當加入PI3K的各種抑制劑(200nM渥曼青霉素、2μMLY294002、2nM咖啡因)完全阻礙S473的磷酸化,也就是說ATM-PI3K-Akt通路中通過抑制PI3K,直接阻礙Akt的磷酸化進行。
4.4 ATM與Akt的下游信號分子S6K1和4E-BP1
在活體雷帕霉素影響的研究中編碼翻譯過程受到許多磷酸化所調控,如核糖體S6蛋白和其激酶p70S6K1以及eIF-4F結合蛋白4E-BP1。Ching JK進行3個單獨實驗驗證ATM在IGF-1刺激下促使S6K1磷酸化下調:①IGF-1刺激的S6K1磷酸化位點在T389,C2C12細胞ATM shRNA中S6K T389磷酸化水平顯著下降;②正常的C2C12細胞(ATM+/+)培育在KU55933中,通過阻礙胰島素通路中的PI3K,進而抑制通路進程來降低S6K1的磷酸化水平;③在觀察只有一半功能的雜合型ATM+/-比目魚肌細胞,在IGF-1刺激下S6K1磷酸化水平顯著低于野生型對照[9]。Burnett PE[12]報告ATM類似家族的相關激酶有助于促進mTOR下游S6K1和4E-BP1磷酸化。Yang DG[82]在研究胰島素刺激下蛋白質合成通路過程中,發現誘導eIF4E的結合蛋白4EBP1,ATM能在4EBP1的S111位點發生磷酸化,當ATM缺失的離體細胞,顯著下調4EBP1從eIF4E分離水平。與此結果相一致的研究Kuang X[83]在ATM-/-小鼠中發現ATM的功能缺失下調mTOR信號通路的4EBP1水平。
5 ATM阻礙蛋白質合成的路徑假設
AT疾病同樣被歸類于惡性體質,會呈現出骨骼肌合成受阻和肌力下降等典型表征[3],雖然AT疾病在世界范圍內的發病率處于1/44 000—1/100 000之間并不高[14],但是自1964年Dunn首次發現至今[42],科學界對其仍然給予極大的熱情[7]。與其他惡性體質的疾病不同,AT疾病是缺少單個蛋白(ATM)的表達[25],導致整體骨骼肌丟失和運動能力等表型下降,因此探索AT疾病與骨骼肌蛋白質合成的關系從實驗的角度來看,就大大提高可行性和可信度。
宏觀視域下,AT疾病與骨骼肌的關系在正文部分較為詳細的論述,AT患者的身高、體重和BMI顯著低于對照組,骨骼肌的體積、橫截面和肌力顯著下降,并且隨年齡增長而漸進式加劇骨骼肌各項表征下降進而影響行走、站立等運動能力[8, 15, 35]。微觀視域下,骨骼肌蛋白質mTOR通路是蛋白質合成的主要信號通路,本文梳理ATM與蛋白質合成mTOR通路中的關鍵分子的相關性,而且所有的結果一致認為ATM阻礙相關分子的蛋白質合成(即負向調控合成)[9],基本構建出ATM阻礙/抑制骨骼肌蛋白質合成的可能路徑(見圖3)。ATM缺失/下調可能發生的假設路徑:下調ATM→下調IGF-1→下調IGF-1R→下調IRS-1(通過下調Y612或上調S307)→下調PI3K→下調Akt(通過下調S473和T308)→下調mTOR→下調S6K1和下調4E-BP1的分離率→骨骼肌蛋白質合成。
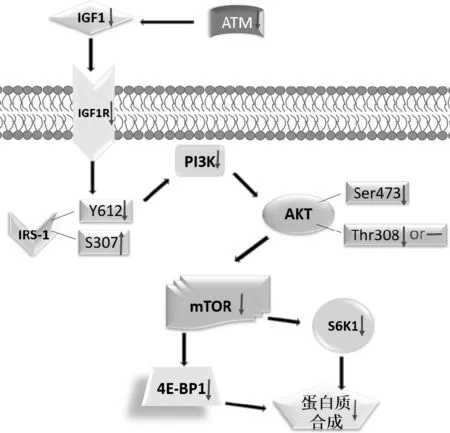
圖3 ATM阻礙骨骼肌蛋白質合成的路徑假設
不同肌纖維對胰島素刺激下的ATM的作用有顯著性不同(脛骨前肌的Ser473磷酸化,而比目魚肌未發生)[72],而且還發現脛骨前肌的ATM蛋白水平是比目魚肌中的10倍,表明ATM更傾向于快肌(脛骨前肌vs比目魚肌)[9]。抗阻運動又是提升骨骼肌中IGF-1的方式,有氧運動并未發現能促進IGF-1的分泌[51],另一項研究證明AT病人經過呼吸肌的鍛煉,力量得到顯著提升[32]。抗阻訓練對ATM蛋白缺失的個體有正向調控,并且提示可作為改善AT疾病病況和提升生活質量的非藥物手段,因此,需要有更多的動物活體實驗通過抗阻運動的干預,探索干預后AT疾病表征和骨骼肌蛋白質合成中ATM蛋白的有益作用。
猜你喜歡
人人健康(2023年26期)2023-12-07 03:55:46
家庭醫藥(2019年9期)2019-09-23 18:54:32
中國生殖健康(2019年2期)2019-08-23 08:12:10
家庭科學·新健康(2018年8期)2018-10-30 10:23:20
人生與伴侶·共同關注(2018年5期)2018-08-15 10:00:00
科學生活(2016年9期)2016-10-20 13:12:45
中國衛生標準管理(2015年1期)2016-01-14 03:41:27
人人健康(2015年17期)2015-09-09 16:25:20
藥學與臨床研究(2015年4期)2015-06-05 11:35:51
中國醫藥科學(2015年15期)2015-02-27 12:32:27
