抗γ-氨基丁酸B受體腦炎患者12例臨床特征分析
2019-01-22 02:09:00戴俊杰章殷希
中國全科醫學 2019年2期
關鍵詞:癲癇
戴俊杰,章殷希
自身免疫性腦炎是一類與神經元表面或突觸蛋白抗原相關的疾病,近年來越來越受到臨床醫生的關注[1]。其主要表現為局灶性或彌漫性神經系統癥狀,目前已知的相關抗體包括針對N-甲基-D-天冬氨酸(NMDA)受體、富亮氨酸膠質瘤失活1蛋白(LGI1)、contactin相關蛋白2(Caspr2)、α-氨基-3-羥基-5-甲基-4-異惡唑丙酸(AMPA)受體、γ-氨基丁酸B(GABAB)和甘氨酸受體等抗體[2]。自身免疫性腦炎最常見的臨床特征包括認知功能障礙、行為紊亂、意識改變和癲癇發作。與典型的神經系統副腫瘤綜合征不同,其常對免疫治療有效,且整體預后較好,但容易復發。部分自身免疫性腦炎好發于兒童和青年,且惡性腫瘤的伴發率與特定抗體相關,一般不會超過70%[1-2]。抗GABAB受體腦炎是一種近期新發現的自身免疫性腦炎,常進展為邊緣性腦炎(limbic encephalitis,LE)[3]。其最典型的臨床表現為早期明顯的癲癇發作(發生率高達80%)[2]、記憶力減退、精神異常、幻覺和性格改變[4]。約半數患者可伴發腫瘤,主要為小細胞肺癌(small cell lung cancer,SCLC)或肺部神經內分泌腫瘤[1,4-5]。全面的免疫調節治療一般可以獲得較好的預后且通常不再復發[2,6]。目前,國內關于抗GABAB受體腦炎的報道相對較少,以個案報道居多[7]。本研究對12例診斷為抗GABAB受體腦炎患者的臨床特征、實驗室檢查、影像學及腦電圖表現、治療和隨訪進行闡述,以期為臨床醫生診治提供參考,提高對本病的認識。
1 資料與方法
1.1 病例資料 選取2012年12月—2017年6月于浙江大學醫學院附屬第二醫院神經內科和寧波市醫療中心李惠利醫院神經內科就診的表現為無明顯病因的急性或亞急性腦炎癥狀的成年患者,共確診12例抗GABAB受體腦炎患者。
1.2 診斷流程 患者均接受以下診斷流程:(1)分析患者首發或主要臨床表現,初篩表現為癲癇發作、精神行為異常、認知功能或記憶障礙的急性或亞急性腦炎。(2)評估患者顱腦磁共振成像(MRI)(1.5/3.0 T)、腦電圖(普通腦電圖或長程視頻腦電圖)、腰椎穿刺腦脊液檢測結果,并對血清腫瘤標志物、血清抗核抗體譜、甲狀腺抗體、胸部CT、腹部及盆腔彩超/CT結果進行評估。(3)獲取患者家屬或本人同意后,進行針對性相關抗神經元抗體篩查。采用基于細胞的間接免疫熒光法(indirect immunofiuorescence,IIF)檢測血清和/或腦脊液中的自身抗體(抗NMDA受體抗體、抗GABAB受體抗體,抗AMPA受體抗體,抗LGI1和Caspr2抗體)和典型的副腫瘤抗體〔抗Yo、Hu、Ri、CV2、兩性蛋白和人抗旁瘤抗原Ma2(PNMA2)抗體〕。稀釋后的標本與用含有人靶基因序列的質粒(Euroimmun AG,Lu?beck,Germany)轉染的人胚腎293細胞反應,異硫氰酸熒光素(FITC)標記的抗人IgG作為第二抗體。根據陽性和陰性對照的細胞質免疫熒光強度確定陽性和陰性反應。抗GABAB受體腦炎診斷依據[7]:(1)急性起病,進行性加重;(2)臨床表現符合邊緣性腦炎;(3)腦脊液淋巴細胞分數輕度升高或者白細胞計數正常;(4)顱腦MRI示雙側或者單側顳葉內側信號異常,或者未見異常;(5)腦電圖異常;(6)血清和/或腦脊液抗GABAB受體抗體陽性。
1.3 治療及隨訪評估 患者均接受免疫治療,并對癲癇發作、精神癥狀進行對癥治療,并予相應支持及康復治療,確診腫瘤患者建議其進行針對性腫瘤治療。通過改良Rankin量表(mRS)評估治療效果和預后:0分為完全緩解,1~2分為顯著緩解,>2分為部分緩解,治療或隨訪后mRS評分不變、增加或死亡為無效。mRS≤2分定義為預后良好,>2分定義為預后不良。
1.4 觀察指標 回顧性收集12例抗GABAB受體腦炎患者的臨床特征、實驗室檢查、影像學檢查、腦電圖表現、治療和隨訪。
2 結果
2.1 臨床特征 12例抗GABAB受體腦炎患者中,男10例,女2例;發病年齡47~71歲,中位發病年齡56歲,主要為中年(45~59歲)患者(9/12);病程為6~120 d,中位病程14 d。4例患者有前驅癥狀(發熱、頭痛、咳嗽、上呼吸道癥狀等)。癲癇發作(9/12)是最常見的初發癥狀,且在所有患者病程中出現;11例患者有不同程度的認知功能障礙,1例患者無法評估;10例患者存在精神癥狀(如幻覺、行為、情緒及個性改變等);10例患者出現意識障礙(見表1);1例患者合并心律失常。
2.2 實驗室檢查 12例患者常規血液學檢測無明顯異常。腦脊液檢查示,12例患者均有不同程度的異常表現。10例患者腦脊液壓力在參考范圍內〔參考范圍:80~180 mm H2O(1 mm H2O=0.009 8 kPa)〕,1例患者輕度升高,1例無相關資料;7例患者腦脊液白細胞計數升高〔參考范圍:(0~8)×106/L〕,5例以淋巴細胞分數增多為主;6例患者腦脊液蛋白升高(參考范圍:15.0~45.0 mg/dl),腦脊液葡萄糖及氯化物均未見明顯異常(見表2)。患者腦脊液培養、腦脊液病毒檢測(單純皰疹病毒、EB病毒和巨細胞病毒)和梅毒檢測結果均為陰性。
12例患者血清和/或腦脊液標本中抗GABAB受體抗體均為陽性。6例患者行副腫瘤抗體(抗Yo、Hu、Ri、CV2、兩性蛋白和PNMA2抗體)的檢測,結果均為陰性。另有7例患者存在其他抗體:1例合并抗NMDA受體抗體陽性,2例合并抗核抗體陽性,2例合并抗β2糖蛋白1抗體陽性(其中1例還合并抗心磷脂抗體陽性),2例合并抗甲狀腺球蛋白抗體陽性(見表2)。
2.3 影像學檢查 12例患者均接受顱腦MRI檢查,其中7例未見明顯特異性異常,5例患者的異常影像主要局限于顳葉內側和海馬,3例患者雙側皆受累(見表2),特征性表現為T1加權成像(T1WI)低信號,T2加權成像(T2WI)/液體衰減反轉恢復序列(FLAIR)高信號,彌散加權成像(DWI)未見彌散受限(見圖1)。1例患者可見鄰近腦膜強化。1例患者行磁共振波譜(MRS)檢查,可見N-乙酰天冬氨酸(NAA)降低和乳酸峰升高。
12例患者均通過胸部CT、腹部和淋巴結超聲檢查篩查潛在的惡性腫瘤。5例患者(病例1、4、7、10、11)可見肺部腫塊,經病理學診斷均為SCLC;1例患者(病例5)CT可見縱隔腫塊,經活檢證實為胸腺瘤B型;1例患者(病例8)正電子發射計算機斷層顯像(PET-CT)提示全身骨代謝高信號,1例患者(病例12)鱗狀上皮細胞癌抗原升高,2例患者均懷疑腫瘤,但拒絕進一步檢查(見表2)。
2.4 腦電圖表現 12例患者均接受腦電圖檢查,10例患者腦電圖可見異常,其中2例患者主要表現為慢活動增多,可局灶性累及顳區,8例患者呈彌散分布,2例患者可見顳區癇樣放電(見表2)。
2.5 治療及隨訪 住院期間,患者急性期均接受一線免疫治療,其中10例患者接受皮質類固醇和免疫球蛋白聯合治療,而病例4和病例7分別單獨使用免疫球蛋白和皮質類固醇治療。合并SCLC的5例患者,2例接受化療,3例放棄手術、化療或放療;合并胸腺瘤的1例患者接受手術(見表3)。

表1 12例抗GABAB受體腦炎患者主要臨床特征Table 1 Clinical features of patients with anti-GABAB receptor encephalitis

表2 12例抗GABAB受體腦炎患者的實驗室檢查、影像學及腦電圖檢查結果Table 2 Laboratory examination,imaging and EEG results of patients with anti-GABAB receptor encephalitis
隨訪0.5~20.0個月,隨訪期間5例患者癥狀完全緩解或顯著緩解,3例患者部分緩解,4例患者癥狀無明顯變化或死亡(其中2例確診SCLC,另外2例懷疑腫瘤),死亡原因均為嚴重肺部感染、呼吸衰竭。未發現腫瘤及已對腫瘤行針對性治療患者預后相對較好(共7例,5例完全或顯著緩解,1例部分緩解,1例無反應、死亡),確診或懷疑腫瘤而未行腫瘤治療患者預后較差(共5例,2例部分緩解,3例無反應最終死亡)(見表3)。
3 討論
雖然自2010年以來已有100多例抗GABAB受體腦炎的病例,但來自亞洲的報道較少[3,5-9]。本研究共收集12例抗GABAB受體腦炎患者的病例資料。
GABAB受體是G蛋白偶聯受體,在神經遞質傳遞和突觸穩定中發揮重要作用。其通過激活G蛋白偶聯的內向鉀通道和抑制鈣通道來介導預激活抑制[10]。突觸后GABAB受體通過相似的機制介導抑制,也誘導緩慢的抑制性突觸后電位[3]。GABAB受體廣泛分布于中樞神經系統,但最常見于大腦皮質、海馬、丘腦和小腦[11],主要參與學習、記憶和認知功能[12]。動物實驗研究表明,如果缺乏GABAB受體(如遺傳障礙或藥物引起),小鼠會出現癲癇和記憶、學習和行為障礙[13]。因此可以推測,抗體結合GABAB受體后可能出現類似的癥狀。
本研究提示,抗GABAB受體腦炎臨床上主要表現為急性或亞急性起病的癲癇發作、認知功能障礙、精神行為異常和意識改變,常見于中老年男性,其中癲癇發作是最早和最突出的癥狀,這與先前文獻描述大致相同[7,14]。事實上,癲癇發作被認為是抗GABAB受體腦炎的關鍵特征之一,主要表現為全面性強直-陣攣發作,抗癲癇藥物治療效果欠佳,可快速進展為癲癇持續狀態[7,14]。本研究中病例1從起病到明確診斷經過了120 d,確診病程較長,細究其病史,其初發癥狀癲癇發作程度較輕,且顱腦MRI未見明顯異常,當地醫院診斷“隱源性顳葉癲癇”門診隨診,過程中常規多種抗癲癇藥物治療效果欠佳,且發作逐漸頻繁、程度加劇,送至本院進一步完善檢查方才確診。提示對于新發藥物難治性癲癇患者要考慮到本病的可能。
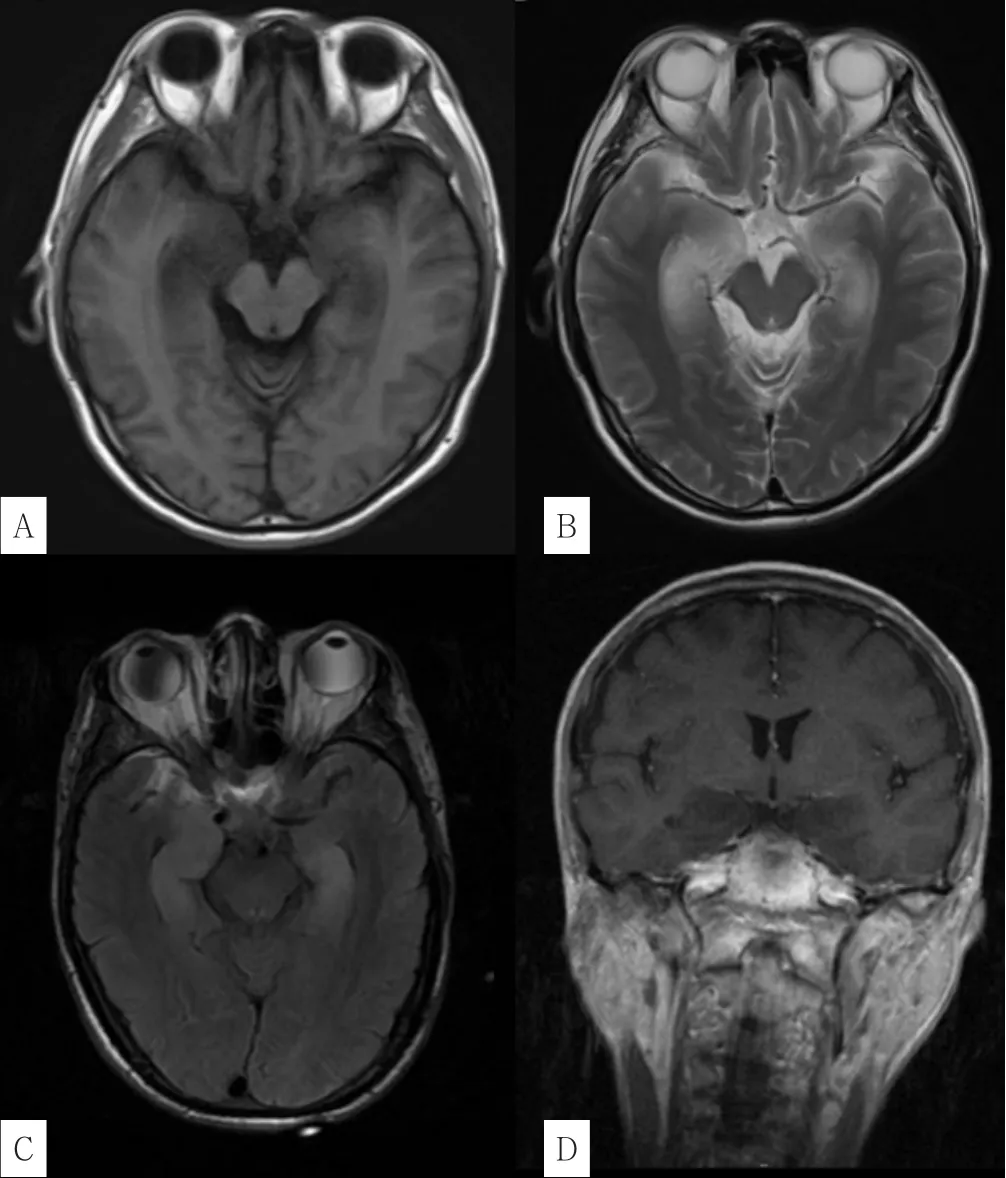
圖1 抗GABAB受體腦炎患者的典型MRIFigure 1 Typical MRI features in patients with anti-GABAB receptor encephalitis
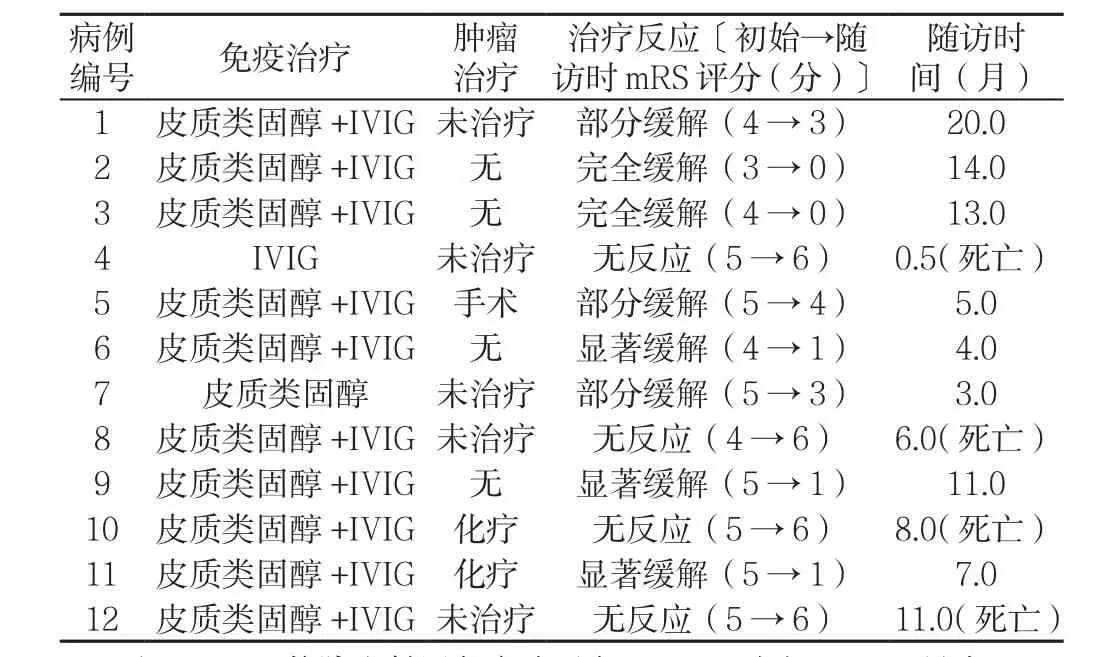
表3 12例抗GABAB受體腦炎患者治療措施及結果Table 3 Treatment responses and follow-up outcomes of patients with anti-GABAB receptor encephalitis
影像學檢查5例患者顱腦MRI可見病灶,其中4例表現為雙側或單側顳葉內側和海馬信號改變(T1WI低信號、T2WI/FLAIR高信號),與大多數研究結果[8-9,14]一致;另1例表現為顳側水腫,易與病毒性腦炎混淆。1例患者的顱腦MRI增強掃描顯示相鄰腦膜增強,這在之前極少被描述。此外,胼胝體和白質病變罕見受累,但也有個案報道[15]。總而言之,顱腦MRI檢查結果陽性率并不高,且并未能發現屬于抗GABAB受體腦炎的特異性表現,也就是說,即便顱腦MRI結果陰性也不排除本病可能。與此相反,腦電圖檢查陽性率明顯更高,雖然缺乏類似于抗NMDA受體腦炎患者中δ刷的特異性表現[7],但大部分患者可出現慢活動增多,少部分患者還可有輔助定位意義的癇樣放電。本研究顱腦MRI陽性患者腦電圖均為陽性表現,但反之不然,考慮原因為抗GABAB受體腦炎患者起病初僅有功能障礙未出現結構改變[9],同時需關注顱腦MRI陰性患者的影像學隨訪,如本研究中病程最長的患者顱腦MRI異常是在起病120 d后復查發現。
盡管GABAB受體已經被證明是神經元表面抗原,而不是典型的細胞內腫瘤神經抗原,但其與腫瘤的相關性仍值得關注。約50%的抗GABAB受體腦炎患者最終發現合并SCLC或神經內分泌腫瘤[1]。本研究12例患者中5例患SCLC,1例肺腫瘤指標增高,1例全身骨代謝高信號,還有1例被確定合并胸腺瘤(之前僅有1例報道[16])。既往也有描述合并罕見的腫瘤類型,包括乳腺癌、直腸癌、多發性骨髓瘤、胸腺類癌、黑色素瘤和食管癌[15,17-18]。因腫瘤的發現常在LE診斷后[4],故對于老年人(特別是有吸煙史者)一旦確診抗GABAB受體腦炎,肺癌的篩查尤為重要。長期隨訪,定期復查腫瘤也是必需的,即使在發病時沒有發現腫瘤。有學者提出,如果腫瘤篩查最初是陰性的,可在3~6個月后復查,并在最開始4年內每6個月復查一次[19]。
現普遍認為,抗GABAB受體腦炎是一種自身免疫性疾病,部分患者血清檢測可能會顯示其他自身抗體陽性,最常見的如谷氨酸脫羧酶抗體、甲狀腺過氧化物酶(TPO)抗體,SOX抗體和抗Hu抗體[3,5-6,20-21]。這些抗體可能很好地反映了疾病與自身免疫或潛在腫瘤的相關性,如與SCLC相關的SOX1抗體[4]。既往報道的抗GABAB受體腦炎合并副腫瘤抗體的比例為 7%~40%[3,5,17,21]。兩種抗體共存主要見于 SCLC 患者中,常提示預后不良[5]。本研究患者行副腫瘤抗體檢測者結果均為陰性,但存在其他自身抗體陽性,盡管未見相應靶器官受累,1例同時合并抗NMDA受體抗體陽性。盡管如此,仍然建議抗GABAB受體腦炎患者均應行副腫瘤抗體檢測,有助于預后的判斷。
目前尚無抗GABAB受體腦炎的統一診斷標準[7],最終主要依賴于抗GABAB受體抗體的檢測,但抗體的檢測費用昂貴且耗時長,甚至有些患者臨床表現符合自身免疫性腦炎表現,但抗體檢測為陰性[22-23]。因此,對于哪些患者進行自身免疫性神經元抗體的送檢、及時啟動免疫治療和如何解決抗體檢測陰性困惑成了臨床的疑難問題。GRAUS等[23]提出了自身免疫性腦炎的可能(possible)、擬診(probable)和確診(definite)標準,這也為抗體陰性的自身免疫性腦炎的診斷提供了依據。此外,針對如同本研究中病程最長的個例患者,近年來提出了“自身免疫性癲癇”的概念[22],DUBEY等[24]也提出了進行相關抗體檢測及免疫治療反應的評分系統,這也為臨床實踐提供了更多的可操作性。
免疫療法普遍被認為是抗GABAB受體腦炎的一線方案[7]。皮質類固醇、靜脈注射用免疫球蛋白和血漿置換可以單獨或聯合應用。既往研究表明,初次發病時接受免疫治療的患者比不接受免疫治療的患者有更好的預后和更低的病死率[25]。與傳統的細胞內抗原抗體相關的LE(例如抗Hu)相比,抗GABAB受體腦炎對治療反應更好[3,5]。本病引起的癲癇發作常為藥物難治性,但通常在免疫治療后效果良好。一般建議在患者血清或腦脊液中檢測到抗GABAB受體抗體后開始啟動治療[3,5,20],但對于抗體檢測陰性或癲癇發作嚴重的患者,臨床上高度懷疑自身免疫性腦炎,可依據GRAUS等[23]和DUBEY等[24]提出的操作流程及早進行免疫治療,而不用過分關注抗體檢測結果,在一線免疫治療效果欠佳時,需重點關注腫瘤的篩查及治療,并可通過免疫治療反應評分系統進行啟動二線治療的決策。
本研究患者中,4例未發現腫瘤患者經免疫治療后均完全緩解或顯著緩解,而發現或懷疑腫瘤的8例患者中,3例部分緩解,1例顯著緩解,4例對治療無反應且最終死亡,提示未發現腫瘤的患者預后相較于合并腫瘤患者更好;而同樣罹患腫瘤患者中,接受腫瘤治療患者(共3例,1例無反應死亡,1例部分緩解,1例顯著緩解)比未采取針對性處理者(共5例,3例無反應死亡,2例部分緩解)預后可能更好。因此,盡管SCLC與預后不良有關[5],仍建議這些患者積極治療腫瘤,以獲得更好的結局。本研究樣本量較小,需要更大樣本量的數據和長期隨訪來明確本病的最佳治療方案。
總之,抗GABAB受體腦炎是一種相對少見但可治療的自身免疫性疾病。對于主要表現為LE癥狀的中老年男性患者,應將本病納入鑒別診斷。而對于常規抗癲癇藥物治療效果欠佳患者,起病階段雖然未見嚴重癲癇發作,若同時合并精神、認知功能障礙,且隨著時間推進發作愈加頻繁或嚴重,也需考慮到該病可能。抗GABAB受體腦炎常合并SCLC,故副腫瘤抗體和腫瘤的全面篩查至關重要。神經科醫師應該熟悉本病的臨床特征,便于及早做出恰當的診斷,有助于及時啟動免疫調節和可能的腫瘤治療,更好地改善患者預后。
作者貢獻:戴俊杰、章殷希進行文章的構思與設計,研究的實施與可行性分析,數據收集,負責文章的質量控制及審校,對文章整體負責,監督管理;戴俊杰進行數據整理,結果的分析與解釋,撰寫論文,論文的修訂。
本文無利益沖突。
猜你喜歡
中國民間療法(2021年5期)2021-06-09 09:21:04
中華養生保健(2020年2期)2020-11-16 00:49:00
解放軍醫學院學報(2020年12期)2020-03-29 05:11:46
中成藥(2017年6期)2017-06-13 07:30:35
飲食科學(2017年5期)2017-05-20 17:11:53
臨床醫藥文獻雜志(電子版)(2017年11期)2017-05-17 04:48:10
安徽醫科大學學報(2015年9期)2015-12-16 11:09:44
中國當代醫藥(2015年7期)2015-03-01 02:01:13
西南軍醫(2015年4期)2015-01-23 01:19:30
西部中醫藥(2014年6期)2014-03-11 16:07:47
