費托合成中碳載體負載鐵催化劑研究進展
2019-03-22 08:16:40孟凡會
天然氣化工—C1化學與化工 2019年1期
程 楊,孟凡會,李 忠
(太原理工大學煤化工研究所,煤科學與技術教育部和山西省重點實驗室,山西 太原 030024)
費托合成(FTS)反應可以將合成氣轉化為超純液態燃料以及其他化學品[1-4],目前工業上多采用鈷和鐵作為活性金屬制備催化劑。相比鈷催化劑,鐵基FTS催化劑價格更便宜,且CH4副產物較少、烯烴選擇性高[1,3]。此外,鐵基催化劑在高溫下具有優異的水汽變換活性,可以使用低氫碳比的煤基合成氣直接進行反應[2]。
γ-Al2O3、SiO2等氧化物與活性組分 Fe之間存在較強的相互作用,導致Fe物種難以被還原和碳化,因而催化活性不高[3,5]。碳材料具有可觀的比表面積以及可調的孔道結構是理想的載體材料,且與活性組分Fe之間相互作用甚弱,近年來在鐵基FTS反應中被大量研究。
目前碳材料負載型鐵基催化劑主要通過兩種方法制備,一是采用浸漬法將活性組分分散在碳材料載體表面或封裝在載體孔道內;二是先合成出含鐵有機復合物,然后通過碳化處理制備出具有碳包覆或鑲嵌結構的鐵基催化劑,且該類催化劑催化性能優異。鑒于近年來碳材料作為載體在鐵基費托催化劑方面被廣泛研究,本文綜述了近期的研究進展。
1 碳材料載體在鐵基費托合成中的應用
碳材料種類繁多,目前在鐵基FTS研究中被主要應用的有活性炭(AC)、碳納米纖維(CNF)、石墨烯(Graphene)、碳微球(CS)、碳納米管(CNT)及新型介孔碳等載體[6]。碳材料的孔道結構、表面酸堿性以及與活性物種之間的相互作用均可影響活性物種的分散、還原,以及反應物/產物的吸脫附,進而影響催化反應性能。
活性炭比表面積大、孔結構發達、價廉,其含有的灰分組成復雜,對費托反應可能起促進作用,也可能起毒化作用。張春等[7]研究了活性炭種類對Fe/AC催化劑FTS反應性能的影響,結果表明椰殼基AC制備的催化劑其活性和C5+液相產物收率均要高于煤基AC的,這可能是由于煤基AC中的雜質太多影響了FTS反應活性。
CNF、CNT、CS 等純度高、表面性質可調、形貌可控,且與Fe的相互作用較弱,因而在FTS反應中常被作為模型載體來揭示金屬顆粒尺寸、助劑等要素與反應性能之間的內在聯系。De Jong課題組以Fe/CNF為模型催化劑研究了Fe在FTS制C2~C4烯烴(FTO)反應中的尺寸效應和助劑效應。研究發現,碳化鐵顆粒尺寸較小時有利于提高催化活性,但CH4選擇性高[8]。此外,助劑還會影響產物的選擇性和粒徑之間的關系,無助劑時,CH4和C2~C4烯烴選擇性則與粒徑大小無關;存在Na,S助劑時,顆粒尺寸越大 C2~C4烯烴選擇性越高,CH4選擇性越低[8]。Xie等[9]研究了顆粒尺寸、Na及S助劑對Fe/CNF催化劑穩定性的影響,發現引入Na,S加速了Fe物種的碳化并提升了初始活性,但會誘導鐵顆粒長大,進而降低催化穩定性。
熊海峰等以化學沉積法制備的CS為模型載體系統研究了酸處理[10]、氮摻雜[11]、助劑[12]、負載方法[10]等要素對FTS性能的影響,結果表明,硝酸處理可以提升Fe/CS催化劑的性能,且沉積沉淀法制備出的催化劑金屬顆粒尺寸小,活性高。
碳納米管(CNT)獨特的管狀彎曲結構在一定程度上會導致π電子云畸變,并使得管內缺電子而管外富電子。包信和等[13]發現CNT具有的這種獨特電子結構納米空間能夠改變金屬納米顆粒的某些特性,從而可以調節其催化性能。當Fe2O3納米顆粒限域在CNT管內時,可以顯著降低Fe2O3的自還原溫度,并發現限域在管內的鐵物種更易被碳化,從而可提高了FTS活性和C5+烴類收率。
Cheng等[14]以還原氧化石墨烯(rGO)為載體制備了不同K含量的鐵基催化劑,發現FTO催化活性與K含量之間呈現火山型曲線關系,當w(K)為0.5%時催化活性達到最大;而C2~C4烴類產物中烯烴比例卻隨K含量的增大而上升。隨后Cheng等[15]還發現Mg助劑的引入能夠降低CO2選擇性,且在Mg、K協同作用下還能夠顯著增加催化活性,并可保持很高的 C2~C4烯烴選擇性(58.8%)。
介孔碳材料,如CMK-3、介孔碳微球等,具有很高的孔隙率,利用其發達的介孔結構不僅可以提高Fe物種的分散,還可以通過孔道的限域作用抑制Fe物種的燒結。Kang等[16]以CMK-3為載體通過熔體浸滲(melt-infiltration)法制備出高分散、高負載量(w=20%)的Fe5C2@CMK催化劑,在FTS反應中表現出優異的催化活性 (FTY 達到510μmolCO·gFe-1·s-1)和 C5~C12組分選擇性(38%)。近期,椿范立等采用溶膠-凝膠的方法合成了球形介孔碳[17]和氮摻雜介孔碳[18],并將其作為載體采用簡單的超聲輔助浸漬法成功合成了負載質量分數超過40%的鐵基負載型FTS催化劑。該策略能夠顯著提高催化劑的金屬負載量,成功解決了一般負載型催化劑由于負載量較低而在低溫條件下FTS反應活性較低的問題。
2 碳材料改性的方法
盡管碳材料具有很多優異的理化性質,但其表面疏水的特性并不利于活性組分的負載和分散,且碳材料與活性物種之間的弱相互作用導致活性組分在高溫時易團聚而失活。為提升反應活性和穩定性,對碳材料載體進行改性處理十分必要。主要的改性方法有酸堿處理、KMnO4表面修飾以及氮摻雜等。
2.1 酸、堿處理
原始碳材料表面含氧基團較少,親水性差,采用氧化性強酸處理可以使其表面官能團化,提高親水性。一般做法是采用濃硝酸加熱回流,且通常所用溫度越高、時間越久其表面官能化程度越高。酸處理后將會引入羧基、羰基和酚羥基等,如圖1所示[6],這些官能團除了能夠增加載體親水性外,還能錨定活性物種。Li等[19]以氮摻雜竹節狀NCNT為載體,研究了不同濃度硝酸處理對Fe/NCNT催化劑FTS反應性能的影響。發現酸化處理后,NCNT的竹節結構被破壞,表面含氧基團增多,Fe物種更傾向落位于NCNT的管內,進而改善Fe的分散并減小顆粒尺寸,且當處理濃度為10mol/L時催化活性最高。
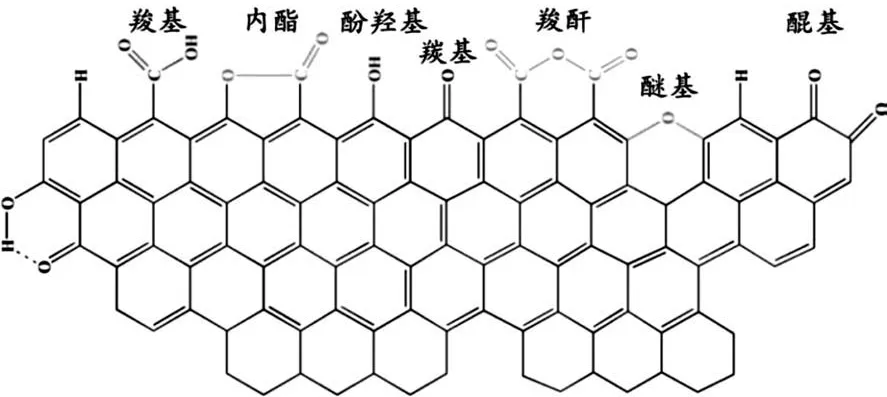
圖1 碳材料表面含氧基團Fig.1 Surfacecontainingoxygen groupson carbon material
近期Liu等[20,21]將堿處理引入到碳材料表面處理中,發現CNT經NaOH堿處理后壁厚減薄,內徑增加,相比于HNO3處理,堿處理的CNT能保持更高的石墨化程度。評價結果表明,用堿處理改性CNT負載Fe,其FTS活性得到大幅地提高,是原始CNT的2.41倍,是酸處理的1.78倍,且堿處理使得費托合成中C10~C20組分選擇性高達60%,遠高于Anderson-Schulz-Flory分布的最大理論值[20]。此外,作者還發現堿處理對活性炭、氮摻雜納米管同樣有提升作用,具有一定的普適性。隨后,又將尿素引入堿處理過程,尿素的引入可以實現氮摻雜,而氮摻雜和堿處理的協同作用使得催化性能得到進一步提高[21]。
2.2 高錳酸鉀表面修飾
眾所周知,K和Mn是Fe基FTS反應中兩種常用助劑,K、Mn助劑能夠促進 C2~C4烯烴的生成、降低CH4副產物,在FTO反應中被廣泛研究。KMnO4是一種含K、Mn的強氧化劑,利用KMnO4與碳基質之間的氧化還原反應不僅可以增加碳材料表面含氧官能團、表面缺陷,還可以在碳材料表面形成K摻雜的MnOx包覆層。
Wang等[22]通過KMnO4氧化修飾CNT得到MnKCNT復合載體,并制備得到Fe/MnK-CNT催化劑。所得Fe/MnK-CNT催化劑鐵顆粒較小、分散度較高,Mn、K助劑與鐵物種微觀尺度混合更均勻,且鐵錳之間的相互作用弱,有利于Fe的還原和碳化,因而催化劑Fe/MnK-CNT催化活性高、穩定性好,低碳烯烴選擇性高(50.3%)。
Tian等[23]以AC為載體制備了系列不同濃度KMnO4修飾的Fe-MnK-AC催化劑并用于FTO反應。結果發現,未經KMnO4修飾的Fe-AC催化劑在反應100h后,活性組分顆粒平均顆粒尺寸從3.2nm增長到26.4nm。而經KMnO4修飾的Fe-10MnK-AC催化劑其顆粒尺寸僅從2.0nm長大到13.5nm,且表現出更高的CO轉化率和C2~C4烯烴選擇性(39.4%)。
2.3 氮摻雜
氮摻雜改性不同于之前的酸、堿處理,其改性方法更加溫和,不會對材料結構產生較大的破壞。利用氮原子的配位錨定作用不僅可以改善活性物種的分散,還可以調變活性物種與載體的相互作用,提高反應活性和穩定性。此外,利用氮原子的富電子特性可以調變碳載體的電子性質,使得部分電子可以從載體轉移到金屬物種上,進而改善催化性能。
包信和等[24]使用氮摻雜石墨烯(NG)為載體,并合成出不同摻雜量的Fe/NG催化劑,發現由于氮原子的富電子性質以及石墨烯極好的導電性,使得NG起到電子給體的作用,增加了Fe周圍的電子密度并促進了CO的解離。在FTO反應中C2~C4烯烴產物選擇性高達50%。
呂金釗等[25]發現,相比AC及酸處理的t-CNT載體,NCNT制備的Fe/NCNT催化劑在FTO反應中表現出更高的CO轉化率和C2~C4烯烴選擇性。作者認為氮摻雜使得載體呈堿性,提高了CO的吸附解離并阻礙了烯烴產物的再吸附和加氫,使得CO轉化率和C2~C4烯烴選擇性都得到了提升。此外,穆斯堡爾譜結果顯示,Fe/NCNT相比Fe/t-CNT在反應中更容易生成χ-Fe5C2活性相。
3 鑲嵌和碳包覆結構新型鐵基催化劑
由于碳材料呈化學惰性且親水性差,一般很難將活性組分均勻分散在載體表面或完全限域在孔道內。盡管碳材料表面改性雖可部分解決此問題,但總體提升效果有限。隨著材料科學的發展,各種新型制備方法涌現,可以直接合成含鐵有機復合物,并通過熱解可以制備出分散均勻和完全包覆的鐵-碳催化劑,而且通常催化性能表現優異。
禹國賓等[26]以葡萄糖和硝酸鐵為原料,通過水熱一步法在溫和條件下得到了碳包覆結構FexOy@C微球。水熱過程中,由于鐵物種的存在加速了糖類的脫水,使得水熱溫度僅需80℃。得益于碳基質的保護作用,在經過108h的FTS反應后鐵物種顆粒僅從7nm增長到9nm,表現出優越的催化穩定性。且無需其他助劑時,C5~C12組分產物選擇性可達到40%。
孫博等[27]通過螯合輔助多組分自組裝的方法制備了有序介孔碳(OMC)負載的鐵基費托催化劑,Fe顆粒尺寸在8.3~22.1nm范圍內可控,制備流程如圖2所示[27]。該催化劑呈現為一種半包覆半暴露結構,Fe納米顆粒鑲嵌在OMC孔道壁中,并暴露于孔道內,這種鑲嵌式的微觀構造使其具有較高的活性和穩定性。且無需助劑時,C5+組分選擇性即可達到68%,遠高于同類催化劑。
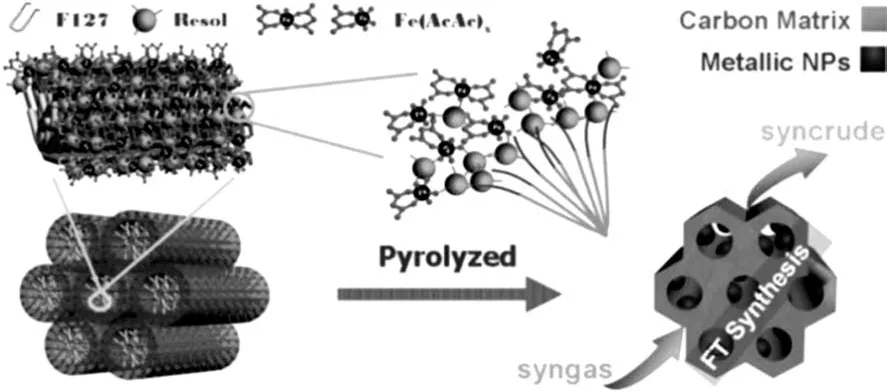
圖2 自組裝方法構造鑲嵌結構Fe-C催化劑Fig.2 Fe-C catalyst with an embedded structure synthesized by self-assembly method
金屬有機骨架材料(MOFs)是一類由有機配體和金屬離子組裝而成的配位聚合物,其骨架結構獨特,且金屬可以以原子級尺度均勻分布[28]。通過碳化MOFs可以得到各種Metal@C材料。Santos等[29]以Fe-BTC(均苯三甲酸配體)為前驅體,糠醇為外加碳源,通過碳化處理制備得到的Fe@C費托催化劑負載量高(w(Fe)=25%~38%),且 Fe物種以極小(2.5~3.6nm)、極高分散的狀態包覆在多孔碳基質中,因而具有超高的活性和穩定性。隨后他們又將Fe-BTC與AlOOH復合,通過熱解制備出Fe@C/Al催化劑,AlOOH的引入提高了C2~C4烯烴選擇性(45%),但是引入過多的Al會導致催化活性降低[30]。
王野等[31]以Fe-MIL-88B(對苯二甲酸為配體)和 Fe-MIL-88B-NH2(2-氨基-對苯二甲酸為配體)MOFs為前驅體,制備了具有Fe3O4@Fe5C2結構的Fe基催化劑,如圖3所示[31]。由于二羧基配體構成的MOFs熱穩定性比均苯三甲酸配體的弱,因而其在熱解時結構遭到更大破壞,金屬納米顆粒尺寸更大(20~30nm)并傾向于分散在碳基質外表面。盡管Fe顆粒較大,但由于暴露了較多的活性位,因而其依然具有很高的催化轉化率。催化劑Fe-MIL-88BNH2/C在300℃、2MPa和空速36000h-1的條件下CO轉化率高達91.8%,且在200h的測試過程中未失活。
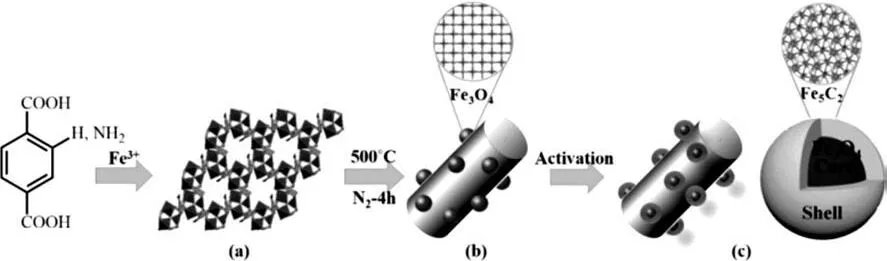
圖3 Fe3O4@Fe5C2催化劑制備示意圖Fig.3 Schematic diagram for Fe3O4@Fe5C2catalyst preparation
4 結論與展望
相比于氧化物,碳材料與Fe之間不存在強的相互作用。碳材料作載體時,不會影響Fe物種的后續還原和碳化,因而反應活性更高,在Fe基費托合成中被廣泛應用。碳材料表面進行改性除了可以調控Fe納米粒子的顆粒尺寸、分散狀態,還能調節Fe與載體之間的相互作用,提高鐵納米顆粒穩定性。新型鑲嵌和碳包覆結構可以阻礙Fe物種的團聚和遷移,極大地延長了其使用壽命,具有很大的研究必要性,定會引發新的研究熱潮。
猜你喜歡
課堂內外·初中版(科學少年)(2025年1期)2025-02-28 00:00:00
課堂內外·初中版(科學少年)(2025年2期)2025-02-28 00:00:00
英語世界(2023年10期)2023-11-17 09:18:18
科學大眾(中學)(2019年3期)2019-05-17 10:04:30
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
汽車觀察(2018年10期)2018-11-06 07:05:26
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
