雙羰基化合成丁二酸(酯)的研究進展
2019-03-22 08:16:40李建國楊先貴王公應
天然氣化工—C1化學與化工 2019年1期
趙 簡 ,孫 騰 ,李建國 ,楊先貴 *,王公應
(1.中國科學院成都有機化學研究所,四川 成都 610041;2.中國科學院大學,北京 100049)
丁二酸又名琥珀酸,是一種重要的有機化工原料及中間體。丁二酸及其衍生物在合成塑料、橡膠、染料、香料、涂料、醫藥、農藥和食品等工業中應用很廣泛,如合成生物可降解塑料聚丁二酸丁二醇酯(PBS)[1-3]。PBS材料具有優異的力學性能和耐熱性能,高度順應世界和國家對環保可降解政策的引導和發展,因此丁二酸具有較好的市場前景[4]。
目前丁二酸的合成方法主要有生物法[5,6](生物轉化法、生物發酵法)、化學法(石蠟氧化法[7]、催化加氫法[8])和電化學電解法[9]。其中,生物法很具有發展前景,但生產成本有待降低;石蠟氧化法雖然工藝較成熟,但是產率和純度都很低,且存在污染。丁二酸在我國的生產起步較晚,前些年我國丁二酸及其衍生產品很多需要進口,近年來隨著國內以微生物發酵為基礎的丁二酸生產工藝的研發與進步,該情況開始改善。國內市場上生產丁二酸的企業有十余家,多數以石油基為主要生產原料,屬高能耗、高物耗、高水資源消耗和高污染的低端產業,且生產規模較小,生產能力相對較低,單線產能最高也僅維持在千余噸左右,這種生產與研發現狀極大地限制了丁二酸的大規模應用[10]。
羰基化反應具有對“原子經濟性”反應的高選擇性和對環境的友好性,可充分利用資源和保護環境,符合綠色化學發展趨勢,備受學術界及工業界青睞[11,12]。雙羰基化反應是通過催化的方法在化合物分子中引入兩個羰基而成為含氧化合物的一類反應[13]。利用乙炔或環氧化物進行雙羰基化反應合成丁二酸(酯)符合國家可持續發展的要求,具有良好的發展前景。
自20世紀50年代以來,人們發展了眾多貴金屬催化體系催化羰基化反應,都取得了良好的催化效果,但是貴金屬價格較高,人們漸漸開始尋找簡單的非貴金屬催化劑代替貴金屬催化劑[11]。本文對丁二酸、丁烯二酸、丁二酸二丁酯和丁烯二酸二丁酯等系列雙羰基化產品的研究情況進行了綜述。
1 乙炔兩步法雙羰基化合成丁二酸(酯)
乙炔可以通過兩步法羰基化合成丁二酸(酯),其反應式為:
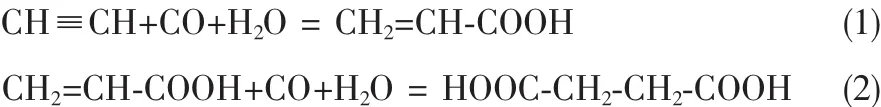
乙炔羰基化法制丙烯酸(酯)是合成丙烯酸的重要方法,1939年,Walter Reppe等首先采用Ni(CO)4催化C2H2、CO和H2O合成了丙烯酸[14],見反應式 (1),之后人們對乙炔羰基化法合成丙烯酸(酯)的研究取得了良好的進展。20世紀60年代,Walter Reppe等[15-17]以丙烯酸及其低級同系物為原料,以Ni、Fe和Co的硫代氰基化合物為催化體系,得到了丁二酸,見反應式(2),他們研究發現活性最好的催化劑為 K2[NiⅠ(CN)3]和 K2[NiⅡ(CN)4]。 他們將282g CH2=CH-COONa、280g K2CO350g K2[NiⅠ(CN)3]加入至1170g H2O中攪拌,在15MPa的壓力下加入CO,然后將高壓釜加熱至150℃,反應24h后,經過蒸餾回收最終得到65g丁二酸。該反應伴隨有副反應發生,反應中會產生多余的CO2和H2。他們還發現:可以以乙炔作為原料合成丁二酸,K2[NiⅠ(CN)3]和K2[NiⅡ(CN)4]能夠同時催化乙炔合成丙烯酸(反應式(1))和丙烯酸合成丁二酸(反應式(2))兩步羰基化反應,且無需分離作為第一步反應產物的丙烯酸。
2 乙炔雙羰基化合成丁二酸(酯)催化劑的研究
20世紀70年代,Chiyuki Fujii等[18,19]采用Pd活性組分-氨基酸催化體系,以C2H2和CO為原料,以醇類為溶劑,在常溫常壓下進行3h雙羰基化反應直接合成了丁烯二酸和丁烯二酸酯,產物中順丁烯二酸二甲酯占90%,反丁烯二酸二甲酯占5%。其反應式為:
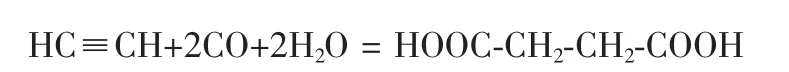
隨后人們以鈀基催化劑為主催化劑,以金屬鹽和酸為助劑,對C2H2和CO雙羰基化一步合成丁二酸(酯)的催化劑展開了研究,結果如表1所示。
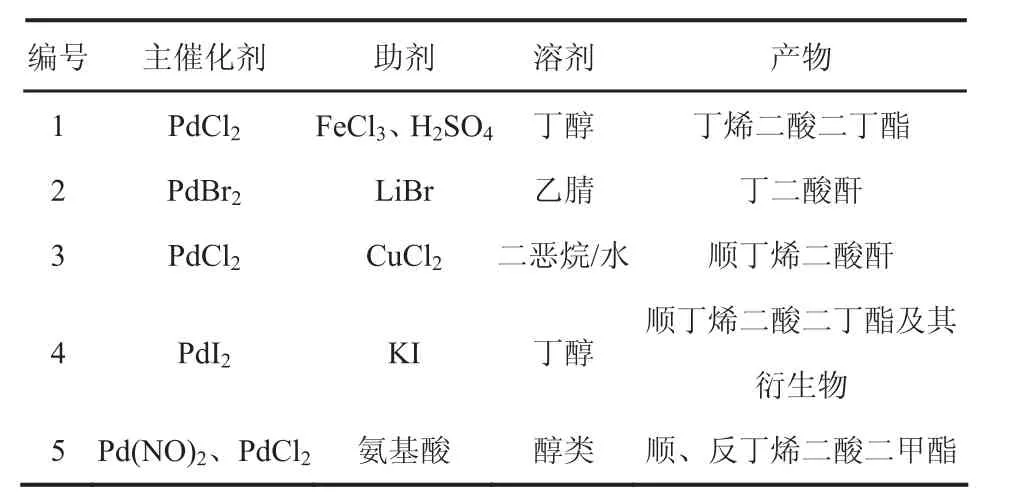
表1 乙炔羰基化合成丁二酸(酯)的鈀基催化體系
20世紀80年代,許松巖、聶宗武等[20-22]采用PdCl2-FeCl3催化體系,以丁醇為介質,探索研究了溫度、壓力以及助劑對合成丁烯二酸二丁酯的反應的影響,研究發現:當催化劑僅用PdCl2單組份時,Pd會很快析出,所得產物極少,催化劑無法循環利用,當加入助劑H2SO4時,能有效地抑制CO2的生成,并可增加丁烯二酸二丁酯的生成量;反應在常壓和60℃下進行時所得丁烯二酸二丁酯的產率為14(gPdCl2·h),收率為 74%,而在 0.6MPa和 80℃下進行時獲得的丁烯二酸二丁酯的產率平均為125g/(gPdCl2·h),收率平均為 84%。
20世紀 90年代,Bruk等[23,24]采用PdBr2-LiBr-CH3CN催化體系,在40℃和101kPa的條件下,乙炔羰基化合成了丁二酸酐,乙炔的轉化率約70%,但此種方法反應速度較慢,還有2,5-呋喃酮、乙烯等副產物產生,后續分離處理較難。
2001年,Jinheng Li等[25-27]用二惡烷和水混合做溶劑,以PdCl2和CuCl2為催化劑體系對末端炔烴進行羰基化反應,以高產率獲得了馬來酸酐。反應式如下:
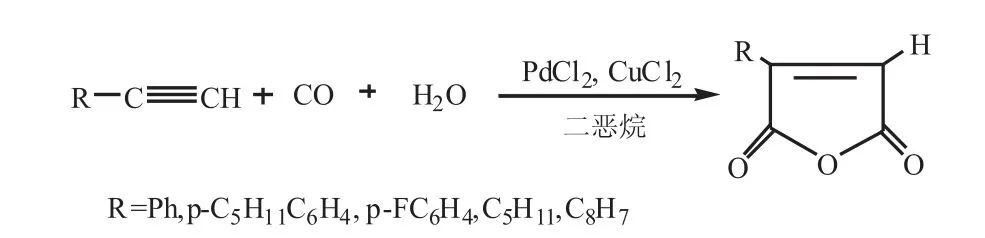
他們研究了助劑對反應的影響,結果表明:①在沒有助劑(例如CuCl2),只用PdCl2作為催化劑的情況下,反應可以平穩進行,但是速率和產率在一定程度上降低,苯乙炔可以在14h后完全轉化為酸酐,反應結束后,催化劑變成黑色固體粉末,X射線晶體學顯該黑色粉末為黑色的Pd單質;②當常溫常壓下, 催化劑為 PdCl2(0.056mmol),CuCl2(10mmol),溶劑為二惡烷(w(H2O)=0.3%)時,向混合物中加入5mmol苯乙炔時,反應能夠順利進行,且馬來酸酐收率達到98%;③當使用PdCl2和CuCl2作為催化劑時,其它末端乙炔也能夠以優異的產率順利地轉化成相應的馬來酸酐;因此,在由PdCl2催化的末端炔烴雙羰基化反應中,助劑不是必要的,但它可以影響反應的產率和速率。
2003年,Bartolo Gabriele等[28-31]使用 PdI2或 PdI2/KI作為催化體系,一步完成了末端芳基乙炔的氫烷氧基羰基化反應,合成順丁烯二酸及順丁烯二酸酐。他們在60~80℃下,以水和二惡烷為溶劑,加入催化劑 PdI2和 KI,在 V(CO):V(空氣):V(CO2)=4:1:10的比例下通入氣體混合反應,得到順丁烯二酸酐收率為48%~96%。
3 環氧化合物雙羰基化合成丁二酸(酯)
由于分子內環張力的存在,環氧化物具有很高的反應活性,環氧化物雙羰基化合成丁二酸(酯)的反應式如下:
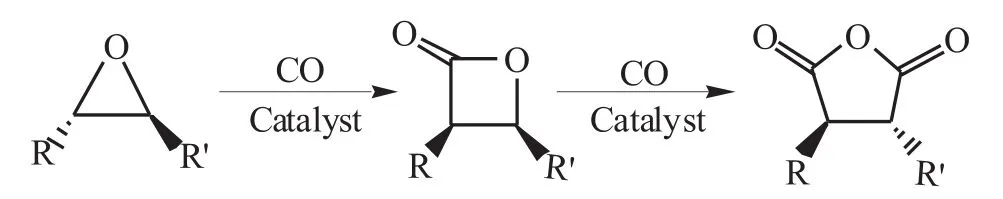
環氧化物先羰基化合成內酯,再由內酯羰基化合成丁二酸酐,最初研究表明:這兩步需要不同的催化劑來完成[32-34]。2006 年,John 等[35,36]研究發現:催化劑[(ClTPP)Al(THF)2]+[Co(CO)4]-(ClTPP=內消旋-四(4-氯苯基)卟啉;THF=四氫呋喃)對該兩個階段都有良好的催化作用,于是成功實現了環氧化物一鍋法雙羰基化合成丁二酸酐,并對催化劑和底物的比例進行了優化,使得1~2mmol的環氧化物在90℃條件下在3h內幾乎完全轉化,丁二酸酐的收率可以達到98%。
2013年,Allen等[37,38]采用連續流動系統,將環氧乙烷與一氧化碳在至少一種路易斯酸與過渡金屬羰基配合物的羧基化催化劑如[(ClTPP)Al(THF)2]+[Co(CO)4]-的作用下進行雙重羰基化得到了琥珀酸酐,并通過結晶將琥珀酸酐從產物流中分離出來,余下的催化劑循環到反應物流中。他們將環氧乙烷、二惡烷和催化劑及CO在壓力為1448kPa、溫度為100℃的混合器中混合,然后調節壓力為1482kPa,溫度為50℃,原料物流進入反應器,產物物流在1378kPa的壓力和90℃的溫度下離開反應器,閃蒸罐回收的氣體中含有體積分數約3%的二惡烷,21%的CO,34%的甲烷和42%的CO2,出口溶液包含質量分數25%的琥珀酸酐。他們研究發現:產物的分離可以通過多種分離手段完成,包括但不限于固液,氣液和液-液分離技術。分離過程分為全部分離和部分分離:全部分離可使幾乎全部反應催化劑保留在再循環物料流中;部分分離可使占反應產物約0%~100%的酸酐產物分離出來,其余未分離的酸酐隨溶劑和催化劑一起參與再循環。
4 雙羰基化合成丁二酸(酯)機理的研究
20世紀80年代,許松巖、聶宗武等[21]對乙炔在PdCl2-FeCl3催化體系下羰基化合成丁二酸二丁酯的反應進行了動力學和機理的討論。他們通過分別考察CO、C2H2和O2的濃度、H2SO4的添加量和催化劑PdCl2的濃度對反應速度的影響,得到了以下動力學方程:
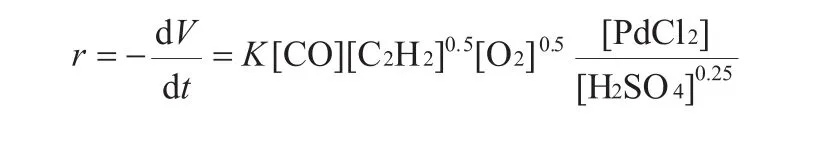
其中:r表示總反應速率,L/min;V表示氣體體積,L;t表示反應時間,min;K 表示速率常數,mol/(L(min)。
20世紀 70年代,Chiusoil等[39]采用 PdCl2-硫脲體系催化乙炔羰基合成丁烯二酸二甲酯和Mekhryahova等[40]采用Pd-膦體系催化合成丁烯二酸酯時,均提出了單核反應機理,但該反應機理不能解釋產物中出現順反異構物的現象。聶宗武等提出的雙核成鍵模型可以很好地解釋產物的構成以及動力學實驗結果,其認為,PdCl2溶液在CO氣氛中可形成不穩定的PdⅠ(CO)Cl絡合物[41],此絡合物在酸性介質中可形成雙核的[Pd2(CO)2Cl4]2-不穩定絡合物,并與C2H2絡合形成具有四面體的 “跨欄”式構型(μ2-η2)的中間物(I),由于部分(I)發生分子間歧化反應而形成“拱橋”式(Ⅱ)與“交叉”式(Ⅲ)中間物;由(Ⅰ)和(Ⅱ)可得順式產物,由(Ⅲ)可得反式產物。 機理如下:
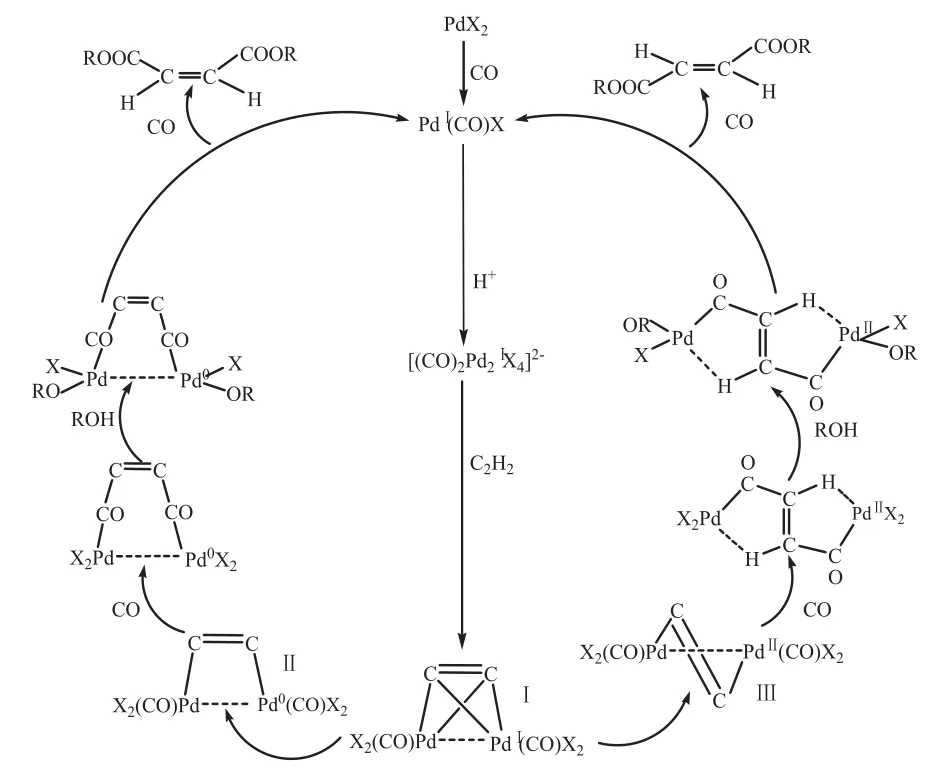
圖1 乙炔催化羰基合成丁烯二酸二丁酯的機理
20世紀90年代,Bruk等[24,35]研究了乙炔在PdBr2-LiBr-CH3CN催化體系下羰基化合成丁二酸酐的反應機理,研究發現:在反應體系中,除琥珀酸酐之外,還產生了較少量的馬來酸酐、琥珀酸、馬來酸、富馬酸和丙烯酸和極少量的丙酸、2-(5)-呋喃酮和乙烯。他們借助專門設計的計算機程序和計算機生成機制的進本反應庫,采用假設演繹方法來研究形成馬來酸酐和琥珀酸的可能機制,其鑒別實驗主要是通過兩個路線:(1)對可能的有機中間體的反應性進行原位研究;(2)通過動力學同位素效應研究和同位素交換反應的研究,從而揭示產生不同的反應之間的動力學關系產品;并在利用原位紅外光譜對中間體進行表征時發現,Pd的氧化態發生了變化,由PdⅡ變成了PdⅠ,催化的活性物質是PdⅠ氫復合物;并通過核磁共振譜圖發現:在乙炔羰基化反應體系中存在鈀氫化物絡合物,而且氫化物配合物的產生最可能是來自于由乙炔的氧化羰基化產生馬來酸酐或馬來酸和富馬酸的過程;他們綜合推測的可能的反應機理如下:
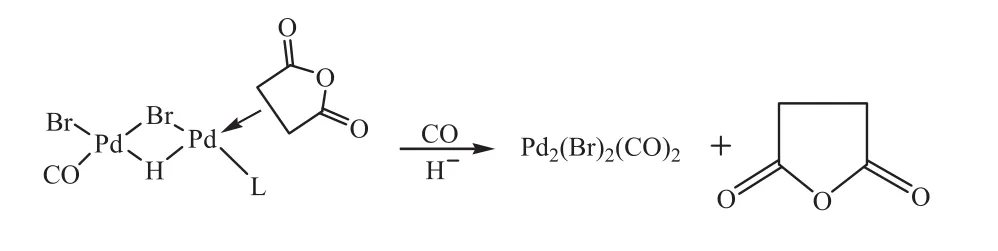
圖2 乙炔羰基化合成丁二酸酸酐的機理研究
2006年,John等[35,36]對環氧化合物雙羰基化合成丁二酸酐進行了催化劑及機理的研究,采用催化劑[(ClTPP)Al(THF)2]+[Co(CO)4]-實現了兩步羰基化的連續反應,他們通過原位紅外光譜研究了該催化劑與環氧化物雙羰基化的機理(圖3),其揭示了兩個羰基化階段是連續的且不重疊的,使得環氧化物羰基化在中間體β-內酯消耗之前完成。
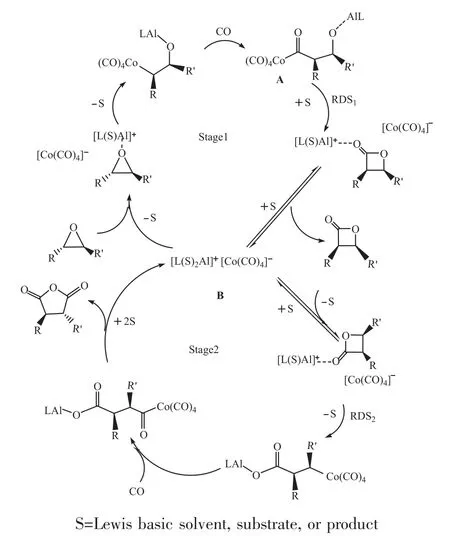
圖3 環氧化物雙羰基化的機理研究
他們分別對兩個階段的機理進行了研究,通過原位紅外光譜監測PO的羰基化時,觀察到在反應過程中γ-丁內酯的形成速率恒定,表明該速率在環氧化物中為零級,又通過原位紅外光譜研究了γ-丁內酯向丁二酸酐的羰基化過程,發現反應速率對整個絡合物的濃度具有一階依賴性。他們發現,不同的溶劑對兩階段的速率影響很大,環氧羰基化速率主要取決于溶劑的給電子能力,而內酯羰基化速率強烈依賴于溶劑的給電子能力和極性[42]。
5 總結與展望
目前,丁二酸(酯)主要是通過石蠟氧化法和催化加氫法進行工業化生產得到,原料大多來源于石油資源。羰基化反應具有高效率、低消耗和低排放的優點,能夠達到可持續發展所要求的生產工藝過程綠色化和資源利用最大化,尤其是利用乙炔雙羰基化制丁二酸(酯)可充分利用我國煤資源豐富的優勢,彌補我國石油資源短缺,為我們提供了一個新的思路。盡管人們對雙羰基化合成丁二酸(酯)的催化劑進行了一些探索研究,但仍存在一些問題,催化劑成本高、催化劑選擇性不高、催化劑壽命和催化劑與產物分離困難等等,今后仍需設計開發高活性、高選擇性和普適性的催化劑體系,并良好實現雙羰基化合成丁二酸(酯)的工業化。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
汽車工程學報(2017年2期)2017-07-05 08:13:02
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
