Al (1 1 1) /Al3Li (1 1 1)的界面性質
2019-04-29 03:08:54孔德斌潘榮凱尹登峰
原子與分子物理學報 2019年2期
孔德斌, 潘榮凱, 尹登峰, ,
(1. 煙臺南山學院工學院,煙臺 265713; 2. 中南大學材料科學與工程學院,長沙 410083; 3. 中南大學,教育部有色金屬材料重點實驗室,長沙 410083)
1 引 言
L12-Al3Li金屬間化合物作為第三代Al-Li合金的重要析出相,能有效改善鋁合金的強度和高溫抗蠕變性能,其時效行為和基本熱力學性能近幾十年來一直鋁合金重點研究領域之一[1-5].研究表明,Al-Li合金的高彈性模量與Al/Al3Li界面特征密切相關[6].因此,研究Al-Li合金析出相(δ′Al3Li等)與基體Al之間的界面性質具有重要的意義.事實上,Baumann, Mao等已經分別從理論和實驗上給出了Al/Al3Li的界面能[7, 8].Gao 等不僅從原子成鍵的角度,計算了Al3Li相的價電子結構[9],而且運用固體經驗電子理論的電子密度分析法,對比計算了δAl3Li和δ′Al3Li相與基體之間的界面電子性質的差異[10],認為Al/δ′Al3Li的界面電子密度在較低應力下保持連續,從而說明δ′Al3Li相與基體結合較好,起到了增強界面的效果.我們認為,研究Al/Al3Li的界面性質,首先應該確定Al和Al3Li形成界面時界面取向, 同一種界面取向,還應該考慮析出相與基體的原子配位關系.同時,基體與Al3Li相的界面附近不同的區域,強度顯然也是不一樣的.界面能只是映了形成界面的難易程度,電子密度分析法也只是間接說明界面區域強度的方法.鑒于此,本文運用密度泛函理論,以Al (111)/ δ′Al3Li (111) 界面取向為例,計算給出了不同的原子匹配關系形成的界面構型.同時用分離功的方法探討最穩定構型的界面附近層與層之間的強度,希望從原子層面反映δ′Al3Li強化合金性能的內在原因.
2 計算方法
所有計算是在基于密度泛函理論的VASP[14]程序中進行的.選擇投影綴加平面波贗勢(PAW)[15]來描述離子—電子間的相互作用,采用廣義梯度近擬(GGA)中的PBE[16]方法處理電子間的交互關聯作用.在計算Al, Li, Al3Li單胞模型以及Al3Li三個低指數面表面時,簡約布里淵區的 K點網格采用Monkhorst-Pack方法(16×16×16)來劃分, 而Al (111) /Al3Li (111) 界面性質的計算則采用8×8×1的K點.所有單胞及表面,界面模型的波函數動能截斷能取320eV.馳豫能量和原子力收斂判居分別為1×10-4和0.02eV/?.
3 結果與討論
首先對Al, Li, 及L12-Al3Li 單胞進行了包括體積和原子坐標在內的幾何優化. 通過 Birch-Murnaghan狀態方程擬合方法得到Al, Li, 及L12-Al3Li 晶格常數 、體模量、基態能(見表1).計算結果與實驗值接近,表明本文采用的計算參數是合理的.
表1Al, Li, 和L12-Al3Li的晶格常數 、體模量、基態能的計算值和實驗值
Table1The calculated results and experimental data of lattice constant, bulk modulus, and ground state energy for Al, Li, and L12-Al3Li.

Elementa/?E0/evB/GPaAl4.041, 4.05 [17]-3.74577.7, 76 [17]Li3.51, 3.44 [17]-1.89913.998, 11 [17]Al3Li4.028, 4.010 [18]-13.5263.91, 66 [18]
本文確定Al (111) /Al3Li (111)的界面模型時,不僅要確定構成界面的表面的原子匹配關系,還要確定構成界面的原子層數,以及真空層的厚度.
不同的結構由于晶格常數的差異,形成界面時,在界面處會產生失配位錯.因此,在構建界面模型時,應該將界面取得足夠大,將界面處的變形均勻地引入其中.但是界面太大,會增加計算量.鑒于此,我們在界面附近的晶格施加了盡量小的應變,使之發生一定的合理變形,以此形成理想的共格界面模型.本文界面處兩側Al和δ′(Al3Li)體模量不同,我們據此設計了不同的變形量.Al(111)面和Al3Li(111)面的應變量分別為壓縮0.11%和拉伸0.22%,應變在界面的方向上是均勻的.
真空層的厚度對界面計算也有影響.厚度太小,原子在Z方向可能馳豫不充分,影響計算結果.厚度太大,會使計算量太大.本文選擇12?的真空層,既可以保持結果準確性,也可以避免計算資源的浪費.
在FCC結構中,Al(111)和Al3Li(111)面都是按照ABC順序堆垛的,因此很容易判斷Al(111)面和Al3Li(111)面有三種配位關系,分別命名為Inter-A, B ,C,如圖1所示.
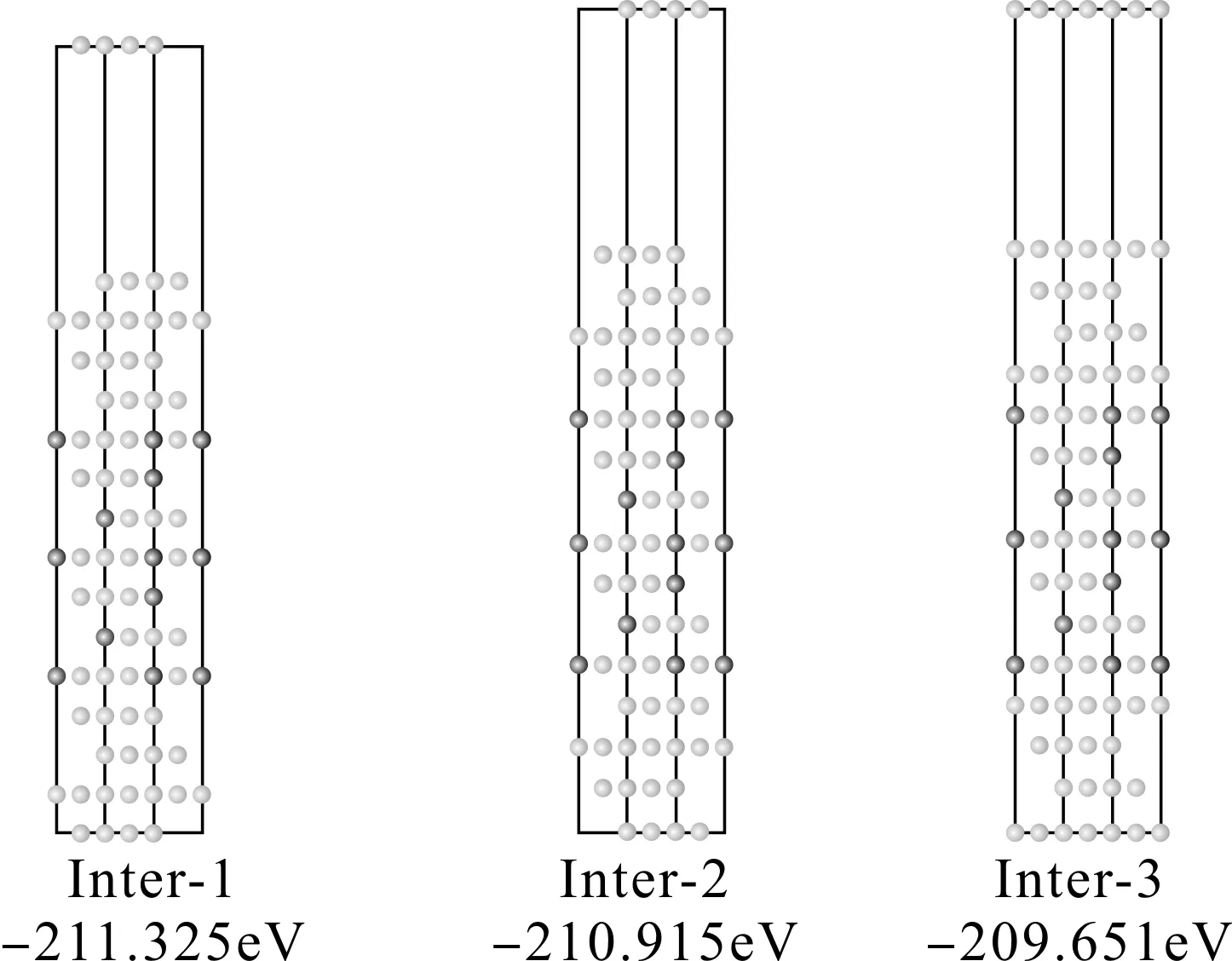
圖1 Al (1 1 1) /Al3Li (1 1 1) 界面三種配位關系示意圖 (白色為Al原子,黑色為Li原子)Fig. 1 Sketch maps of three coordination relations for Al (1 1 1) /Al3Li (1 1 1) interface (blue and green colors represent Al and Li atoms, respectively).
本文對上述三種三明治形的共格Al/Al3Li/Al界面模型分別進行了馳豫計算.整個界面超胞模型包含上下各4層Al原子,中間7層Al3Li和12?的真空層.采取中間層固定,上下層同時馳豫的方法.計算結果表明,以上三種配位關系的界面模型中,Inter-1配位關系的界面超胞總能最低 (如圖2所示),而且馳豫前后界面結構基本保持不變.自由能越低越穩定,所以可以確定Al/Al3Li/Al界面最穩定的配位關系為Inter-1.這是可以理解的.因為Inter-1界面處原子排布仍然采用的是ABC順序堆垛的三明治結構,與基體Al完全一致,對于FCC結構,這種原子排布更有利于結構穩定,所以能量最低.

圖2 Al和Al (1 1 1) /Al3Li (1 1 1) 界面的差分電荷密度(淺色表示電荷增加,深色表示電荷減少)Fig. 2 The deformation charge density Al and Al (1 1 1) /Al3Li (1 1 1) interfaces (yellow and blue colors represent the increase and decrease of the charge, respectively).
為了進一步說明成鍵時界面處的特性與基體Al的差別,我們在圖2給出了Al (111) 面以及Al (111) /Al3Li (111) 三種界面的差分電荷密度圖.可以看出,Al(111)面的層與層之間電荷均勻分布,表現出了金屬鍵的特性.而對于Al (111) /Al3Li (111)三個界面,不僅界面處成鍵特性發生了改變,界面附近區域電荷分布也與基體Al存在差別.不過,相比與Inter-2和Inter-3,Inter-1不論是界面處,還是界面附近,電荷分布與基體Al的分布最為接近,因此其穩定性應該是最好的,這與上文能量計算的結果是一致的.
本文采用界面粘附功Wads和界面分離功Wsep來定量研究界面的結合強度.界面粘附功是兩個自由表面結合成一個界面所放出的能量,結合前后原子都充分馳豫到穩定狀態.界面分離功是界面瞬間斷裂成兩部分所需提供的能量,斷裂后,原子來不及發生馳豫.前者一般可以反映生成界面兩相的潤濕性,后者可以用來評價界面的斷裂強度.一般的材料,斷裂往往發生在分離功最低的地方.但是需要注意的是,分離功衡量的是整個界面區域的強度,不僅僅只反映界面處的結合強度.粘附功和分離功的計算公式定義如下:

圖3 Al(1 1 1)/Al3Li(1 1 1)界面粘附功Fig. 3 Adsorption work of Al (1 1 1) /Al3Li (1 1 1) interface.

圖4 Al(1 1 1)/Al3Li(1 1 1)界面分離功. (1) 界面各層分離功 (2) 界面在最薄弱層處發生斷裂的結果Fig. 4 Separation work of Al (1 1 1) /Al3Li (1 1 1) interface.(1) Separation work for each layer. (2)The results of fracture for the weakest position of the Al (1 1 1) /Al3Li (1 1 1) interface.
本文計算的粘附功為1.588J/m2,但這只是反映了界面結合時的難易程度.為了說明整個界面區域的強度,我們計算了界面處5個可能的斷裂面的分離功,結果如圖4所示.圖4(1)給出了界面可能的斷裂面的位置.需要說明的是,每一個斷裂面的位置都是上下對稱的,我們給出的僅是界面的上半部分.圖4(2)展示的界面在最薄弱層斷裂的結果.可以看出,最薄弱層發生在Al3Li內部,計算結果表明,該處的分離功值是最小的,為1.53eV.而基體Al內部的分離功隨著離界面距離增大,其值逐漸增大.Al3Li內部的各層結合強度表現也出了相似的變化規律.
4 結 論
Al (111) / Al3Li (111) 的界面具有三種原子配位關系結構,界面處仍保持與基體Al相同的三明治堆垛結構的模型,馳豫后能量最低.相比于其它兩種結構,其界面處及界面附近電荷分布與基體Al相比,變化不大.計算表明,該結構強度最弱處位于Al3Li內部.基體Al及Al3Li內部的分離功隨距離界面的距離的增加而逐漸增大.
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
哲學評論(2021年2期)2021-08-22 01:53:34
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
當代陜西(2020年13期)2020-08-24 08:22:02
數學物理學報(2020年2期)2020-06-02 11:29:24
中華詩詞(2019年7期)2019-11-25 01:43:04
制造技術與機床(2017年5期)2018-01-19 02:49:17
濰坊學院學報(2016年2期)2016-12-01 13:00:11
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
光學精密工程(2016年6期)2016-11-07 09:07:19
