Novel analgesic targets and corresponding analgesic leading compounds
2019-06-18 08:14:24ZHENGBoyangZENGRuihong
中國藥理學(xué)與毒理學(xué)雜志 2019年12期
ZHENG Bo-yang,ZENG Rui-hong
(Department of Immunology,Hebei Medical University,Shijiazhuang 050017,China)
Abstract:Opioid medications have been used for pain management,but there are negative side effects,including a potential delay in recovery and increased risk of permanent disability.In this paper,analgesic targets including N-methyl-D-aspartate receptors,cannabinoid receptors,prostaglandin E2 receptor 4 receptors,matrix metalloproteinase receptors,and some new targets for blocking pain signaling pathways associated with these receptors are reviewed.In addition,some novel agonists,antagonists,and leading compounds with agonistic(antagonistic)activities interacting with the target are also described.These novel compounds usually have better analgesic activity and lower side effects than traditional opioids.They are expected to be developed into new analgesics and benefit clinical patients who need pain treatment.
Key words:analgesic;target;agonist;antagonist;leading compound
Pain is a prevalent complex medical problem,characterized by physically debilitating and mentally destabilizing conditions.Current treatment of pain mainly includes non-steroidal anti-inflammatory drugs(NSAIDs),anticonvulsants,antidepressants and opioid analgesics acting on the central nervous system(CNS).The analgesic effects of these drugs do not meet clinical needs and many cause serious adverse reactions.Although opioid analgesics cause powerful analgesic effects,they are associated with respiratory depression,constipation,drug tolerance,addiction and other problems.They are likely to lead to social problems such as drug abuse,thus limiting the clinical use of opioids.These harsh realities call for the discovery of novel analgesic targets and the development of new effective analgesic drugs with lower side effect.Here,we review some recently discovered new analgesic targets and corresponding analgesia leading compounds with agonistic(antagonistic)activities.
1 NMDA receptor related pain pathway and related blocking leading compounds
NMDA receptors are a type of excitatory glutamate receptors in the CNS,which are composed of NR1,NR2(A,B,C,and D),and NR3(A and B)subunits.Studies show that NMDA receptors induce and maintain the central and peripheral sensitization during pain states and visceral pain.The NR2B appears particularly significant for nociception,thus leading to the possibility that blocking agent targeting NR2B and related signaling pathways may be promising in the therapy of chronic pain(Fig.1).Calcium influx through NMDA receptors in the synapse can trigger the phosphorylation of neuronal nitric oxide synthase(n-NOS)adjacent to the ion channels.The phosphorylated n-NOS can synthesize NO and cause the accumulation of NO.The latter upregulates the KATP-mediated currents and accelerates the hyperpolarization of nociceptive neurons.Therefore,blocking the signaling transmission from NR2B to n-NOS to prevent n-NOS activation might be a good way to relieve pain.
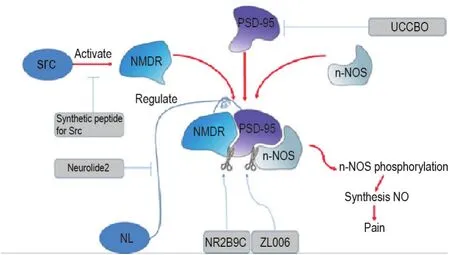
Fig.1 NMDA receptor related pain pathway and related blocking leading compounds.NMDR:N-methyl-D-aspartate receptor;PSD-95:postsynaptic density 95;n-NOS:neuronal nitric oxide synthase;NL:neuroligin.
NMDA receptor antagonists such as dizocilpine MK-801 show obvious anti-nociceptive action in humans and animal models of chronic pain.And NMDA 2B receptor antagonist Ro 25-6981 can attenuate neuropathic pain due to its postsynaptic density 95(PSD-95)expression inhibiting activity[1].But their therapeutic effects are always accompanied by severe disturbances of cognitive and motor functions.Therefore in recent years,many substances acting on NMDA coupled structures have been discovered.
1.1 PSD-95 and related blocking leading compounds
PSD-95 is a medium in the synaptic region where the NR2B and n-NOS are co-localized.It functions as a scaffolding protein that not only attaches NMDARs through its N-terminal PSD-95/Dlg/ZO-1(PDZ)domain but also binds to n-NOS.NR2B9c seems to be an effective analgesic leading compound with minimal side effects.Spinal delivery of the mimetic peptide Tat-NR2B9c disrupts the interaction between PSD-95 and NR2B subunits in the dorsal horn and selectively reduces NMDA receptor-dependent events including wind-up of spinal sensory neurons,and both persistent 40% formaldehydeinduced neuronal activity and pain-related behaviors[2].Tat-HA-NR2B9c can alleviate oxaliplatininduced neuralgia by blocking PSD-95 and NMDAR[3].Intrathecal administration of Myr-NR2B9c can attenuate bone cancer-evoked mechanical allodynia,thermal hyperalgesia,and reduce spinal phospho-Tyr1472 NR2B,n-NOS,and PSD-95 expression[4].This may be a novel strategy for the treatment of cancer associated pain.
PSD-95 inhibitor was also reported to avoid the adverse effects of direct NMDA receptor blocking,because the NR2B receptor activation and calcium influx could still occur.The new compound UCCB01-125 was compared with the NMDA receptor antagonist MK-801 and showed low side effects in the test.UCCB01-144 with more enhanced blood-brain-barrier(BBB)permeability was proved to improve analgesic action[5].Recently,UCCB01-144 was found to reduce reducing neurotoxic PSD-95-mediated signaling and improve neuronal survival following focal brain ischemia in rodents under various conditions and without causing cardiovascular side effects[6].Data suggests that PSD-95 inhibitors involving UCCB01-125 and UCCB01-144 are feasible strategies to prevent both development and maintenance of chronic inflammatory pain and neuropathic pain,and will be used as a leading compound for further optimization in order to develop anti-hyperalgesic agents.
1.2 Src-NMDA interaction and related blocking leading compounds
Src,a type of tyrosine kinase anchored within the NMDAR complex through an adaptor protein,multiples intracellular signaling cascades converge to increase NMDAR activity[7].NADH dehydrogenase subunit 2(ND2)is identified as a Src unique domain-interacting protein.Disrupting the Src-NMDA interaction by ND2 fragment has been reported to be a strategy to treat pain,which not only avoids the undesirable effects of blocking NMDAR function but also avoids directly inhibiting the catalytic activity of Src[8].A team synthesized and screened a series of peptides covering Src40-58 and selected the best ones,Src40-49,with uncoupling activity as the leading compound[9].Later,it was further demonstrated that up-regulation of ND2 played an important role in nociception and pain induced by activation of NMDA receptor after nerve injury[10].Src Homology 2 domain-containing protein tyrosine phosphatase 1(SHP1)interacts specifically with GluN2A subunit of NMDA subtype of glutamate receptors in the spinal cord dorsal horn.Intradermal or interdermal application of GluN2A-derived synthetic polypeptide disrupted GluN2A/SHP1 interaction,alleviated the pain hypersensitivity caused by carrageenan,complete Freund's adjuvant and formalin[11].
1.3 n-NOS protein and related blocking leading compounds
Spinal D-serine modulates n-NOS activity and concomitant NO production leading to increases in PKC-dependent phosphorylation of GluN1(pGluN1)and ultimately contributing to the induction of mechanical allodynia following peripheral nerve injury[12].Choiet al[13]proved that activation of n-NOS could lead to central σ-1R-induced pain hypersensitivity.Two small molecular inhibitors IC87201 and ZL006 directly inhibited binding of purified PSD95 and n-NOS proteins in Alpha Screen[14].IC87201 and ZL006 preferentially suppressed phase 2A pain behavior in the Formalin test and suppressed allodynia induced by intraplantar complete Freund′s adjuvant administration.The group showed that IC87201 administration on post-operative days 0-3 also attenuated the chronic constriction injury-induced development of mechanical all odynia (MA) and pGluN1[15].Then Caiet al[16]expanded the study of ZL006 activity and found that it was effective against hemorrhagic thalamic pain.Another n-NOS inhibitor NG-monomethyl-L-arginine(L-NMMA)was reported to attenuate bone cancer pain,inflammatory and neuropathic pain[17].
1.4 Neuroligin(NL)and related blocking leading compounds
NL1 forms a complex with the presynaptic neurexin-1β (Nrx1β),regulating clustering of NMDA receptors with PSD-95 to underlie pain-related spinal neuroplasticity.Some scholars concluded that NL-induced allodynia mediated by the spinal NL1/PSd-95/pNR2B cascade can be prevented via blockade of trans-synaptic Nrx1b-NL1 interactions.The recombinant Nrx1b Fc chimera and the antisense small-interfering RNA targeting to NL1 may be developed into a new analgesic[18].Interdicting the interaction between PSD-95 and NL 1 or 2 can reduce the synaptic targeting of GluR1 in dorsal horns and in turn inhibit hyperalgesia[19].Besides,targeting of NL2 with the recently discovered peptide inhibitor neurolide 2 can reduce NL2 expression and reverse the established hyperalgesic priming,thus exerting analgesic effect[20].
2 Cannabinoid receptors related pain targets and analgesia leading compounds
There are two subtypes of G protein coupled cannabinoid receptors,CB1 and CB2 receptors(Fig.2).CB1 receptor is mainly distributed in the CNS,including the brain,spinal cord and dorsal root ganglion.CB2 receptor is mainly distributed in immune organs.Both endogenous and exogenous cannabinoid receptor ligands have been shown to mitigate pain[21].Besides,non-CB1R,non-CB2R targets related to the endocannabinoid system have also been reported to be involved in analgesic effect,such as(fatty acid amide hydrolase,FAAH),(monoacylglycerol lipase,MAGL)and(orexin 1,OX1)receptors.
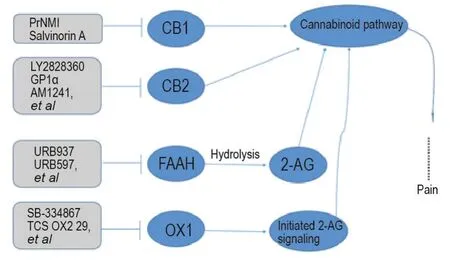
Fig.2 Cannabinoid receptors related pain targets and analgesia leading compounds.CB1:cannabinoid receptors 1;CB2:cannabinoid receptors 2;FAAH:fatty acid amide hydrolase;OX1:orexin 1;2-AG:2-arachidonoylglycerol.
2.1 CB1 and CB2 receptor agonists
Selective CB1 agonists have been studied as a series of novel analgesic mainly for neuropathic pain and inflammatory pain.PrNMI is one compound from a series of recently developed synthetic peripherally restricted cannabinoids,which suppresses chemotherapy-induced peripheral neuropathy pain symptoms by CB1 receptor activation[22].Salvinorin A acts as both a κ-opioid receptor and CB1 agonist to alleviate neuropathic pain[23].Recently,Pandeyet al[24]screened thein silicopurchasable natural products subset of the ZINC12 database against CB1 receptor model and yielded seven compounds as a potent and selective CB1 inverse agonist.LY2828360,a selective CB2 receptor agonist,suppresses chemotherapy-induced neuropathic pain with prolonging effective opioid analgesia while reducing opioid dependence[25].GP1a,which has activity at CB2 receptors,had analgesic activity in the Formalin pain test,and in combination with morphine showed enhanced analgesia[26].AM1241 excites CB2 and sequentially increase morphine antinociception and reduce morphine tolerance and physical dependence[27].AM1710 and MDA7 are another two CB2 agonists which suppress partly chemotherapeutics-induced mechanical and cold allodynia (AM1710)[28]and neuropathic pain(MDA7)[29].GW405833 was originally reported to relieve pain through CB2,but a recent study suggested that its antiallodynic efficacy was CB-dependent but did not seem to involve engagement of the CB receptor′s orthosteric site[30].
Some non-selective CB receptor agonists are useful in pain therapy.WIN55,212-2(WIN),which binds to both CB1 and CB2 receptors,gave synergistic analgesia in combination with morphine in the Formalin test.
2.2 FAAH and related blocking leading compounds
FAAH is a key enzyme in the hydrolysis of the endocannabinoid 2-arachidonoylglycerol(2-AG).FAAH inhibitor can decrease neural excitability and increase signals related to the endogenous cannabinoid pathway in the insular cortex[31],indicating a new target for analgesia,inflammatory pain and neuropathic pain without unwanted side effects typical of direct CB1 agonists.FAAH inhibitors URB937 and URB597 were demonstrated to represent a therapeutic avenue to reduce the overall amount of opioid needed for neuropathic pain,suppressed the development of paclitaxelinduced neuropathic pain and reduced the maintenance of already established allodynia with sustained efficacy without producing tolerance[32].Microinjection of URB597 into the insular cortex causes analgesic effects by decreasing neural excitability and increasing signals related to the endogenous cannabinoid pathway in the insular cortex[31].ASP8477 is another high selective FAAH inhibitor in CNS as analgesia with wide safety margin[33].AGN220653 is a new multitarget compound,with efficacious analgesic and antiinflammatory activities,which are partly due to its capability to achieve inhibition of FAAH and subsequently increasing the tissue concentrations of the endocannabinoid anandamide[34].PF3845,another analogous leading compound,has been reported to suppress neuroinflammation.Bhuniyaet al[35]discovered a novel nicotinamidebased lead 12a through conceptual design and modification of urea moiety in chemotype PF-3845/04457845,which exhibited dose related antihyperalgesic effects.In addition,Kodaniet al[36]designed a dual inhibitor with improved inhibitory potency on both the enzymes FAAH and soluble epoxide hydrolase(sEH)by modifying the carbamoylating and leaving groups from PF-750,a FAAH weak inhibitor of sEH.Full FAAH inhibition with partial MAGL inhibition via co-administration of PF3845 and JZL184 produced a synergistic augmentation of antinociceptive potency.Dual FAAH/MAGL inhibition and selective FAAH inhibition might be preferred for determining the effect of pain management in further studies[37].
2.3 OX receptor,a new analgesia target
Scholars revealed an endocannabinoid-dependent analgesic mechanism mediated by orexin 1 receptor(OX1R)-initiated 2-AG signaling in the ventrolateral periaqueductal gray (vlPAG)[38].Blockade of the OX1R or OX2R in the Cornu Ammonis 1 region of the hippocampus with SB-334867(OX1R antagonist)or TCS OX2 29(OX2R antagonist)decreased the lateral hypo-thalamic-induced antinociceptive responses in the model of orofacial Formalin test in rats[39].Moreover,activating either OX1 or OX2 receptors in the vlPAG can lead to antinociception,but only OX1R-initiated antinociception is endocannabinoid-dependent.Orexin peptides and their receptors have been proposed as new targets for developing analgesic drugs.
3 EP4 receptor and related analgesia leading compounds
Prostaglandin E2 receptor 4(EP4)is a prostaglandin receptor for prostaglandin E2(PGE2).EP4 receptor antagonist can block inflammation pain without the side effects of traditional NSAIDs disrupting the homeostatic function of PGE2.Grapiprant,a selective EP4 receptor antagonist,has also been evaluated in a carrageenan-induced inflammatory pain model in rabbits,which appears to be an attractive option for antinociception in rabbits,due to its rapid onset and extended duration of effect[40].GSK726701A,a novel EP receptor partial agonist,has demonstrated robust activity in a range of pre-clinical models of pain[41].Furthermore,CJ-023423,an antagonist of EP4 receptors,may be a lead compound for the development of novel therapeutics against neuropathic pain caused by injury to the somatosensory system,which is usually refractory to conventional analgesics.
4 Matrix metalloproteinases(MMPs)related analgesia targets and leading compounds
Pain experiments using MMP-9 antagonists showed that MMP-9 antagonists have analgesic activity and can reduce opioid tolerance[42-43].Pretreatment with resveratrol effectively alleviates trigeminal neuralgia in early stages via a powerful inhibition on the activation of MMP-9/2[44].Ronsisvalleet al[45]found that new compound,(-)-MML1017,a μ-opioid receptor benzomorphanic agonist,might elicit analgesia acting as MMP-9 inhibitors.Oral dosing with the dual active,MMP-2/9 inhibitor,AQU-118,attenuated mechanical allodynia in the spinal nerve ligation(SNL)rat model of neuropathic pain[46].
5 Summary
Ten analgesic targets and thirty-three novel corresponding analgesic leading compounds are reviewed(Tab.1).Most of the leading compounds interacting with their targets show obvious analgesic effect with low side effects,which may be novel strategies for the treatment of neuropathic pain,chronic inflammatory pain and cancer associated pain.More studies are needed to develop new analgesics of clinical application value.It is believed that in the near future,more patients will be able to receive pain treatment and use new low-side effect analgesics.
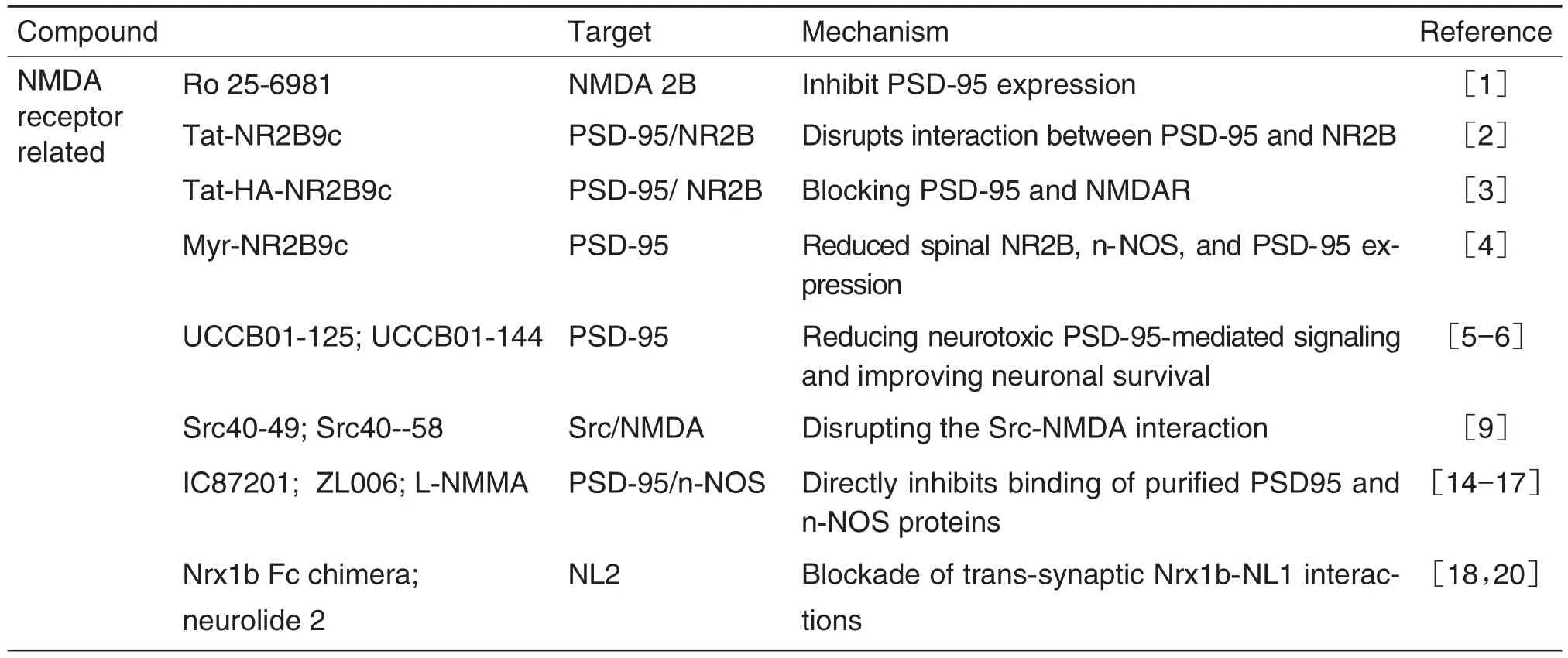
Tab.1 Novel analgesic targets and corresponding analgesic leading compounds
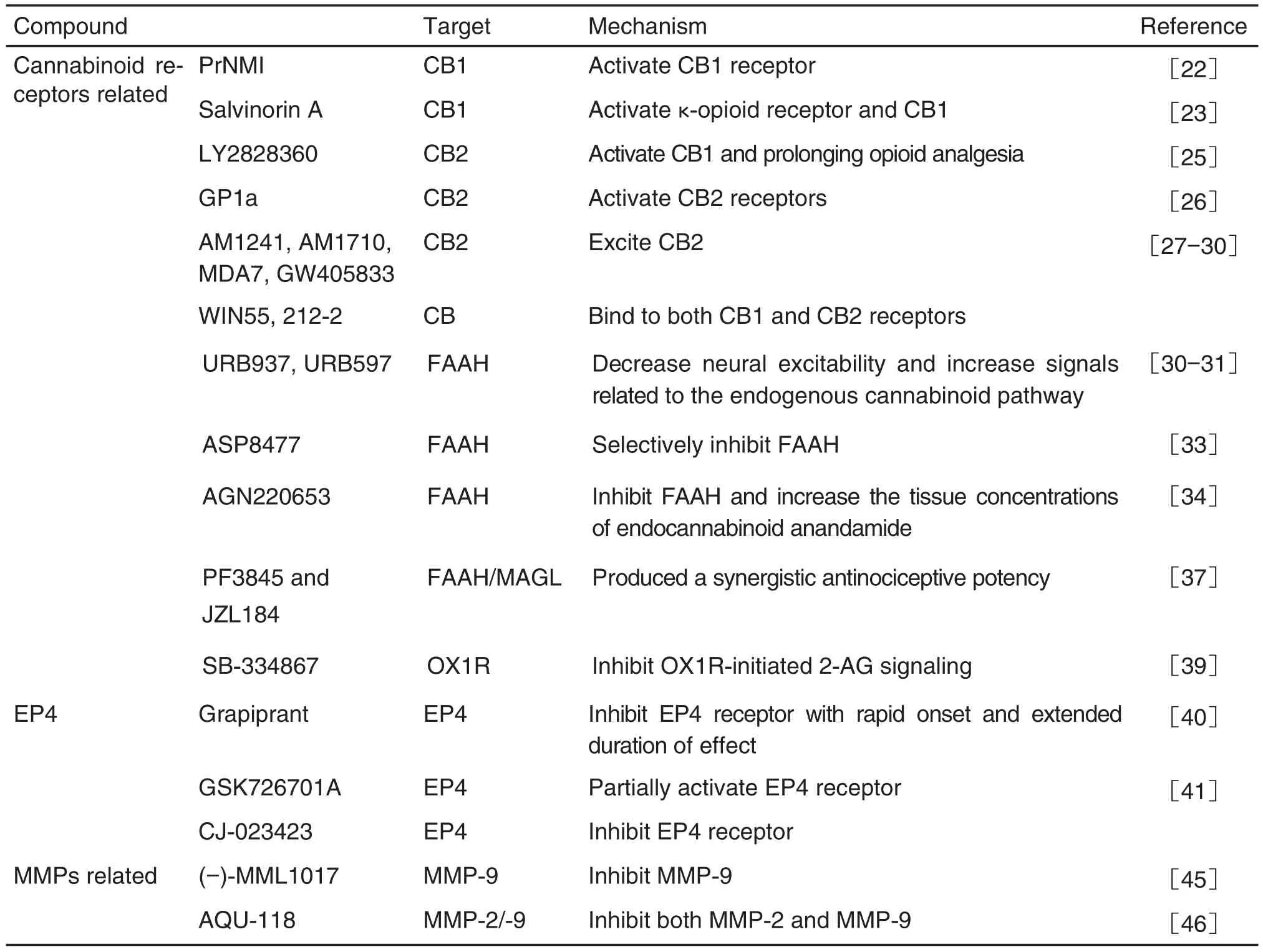
續(xù)表1
