空間位阻凈化-超高效液相色譜-串聯質譜技術測定動物源性食品中抗組胺類藥物殘留
2019-07-05 02:13:34葉佳明鐘世歡陳青俊
食品科學 2019年12期
王 京,葉佳明,王 瀟,鐘世歡,陳青俊
(1.贊宇科技集團股份有限公司,浙江 杭州 310030;2.浙江省輕工業研究所,浙江 杭州 310009;3.浙江公正檢驗中心有限公司,浙江 杭州 311305)
隨著膳食結構的不斷改善和對動物性蛋白質需求的不斷增加,人們對肉制品、奶制品和魚制品等動物性食品的質量要求也就越來越高,對食品的獸藥殘留引起了普遍關注,并認為獸藥殘留將是今后食品安全性中重要問題之一[1-2]。抗組胺類藥物,可用于鎮吐、抗暈眩、暈動癥以及鎮靜催眠。其在獸醫臨床上的藥理作用廣泛,飼料中添加此類藥物,能興奮攝食中樞,不法商人在動物養殖過程中對其非法使用,從而達到增加動物采食和促進生長增加體重等作用[3];另外,使用抗組胺類藥物可降低動物運輸過程中的死亡率。因該類藥物脂溶性高,易蓄積于脂肪組織,停藥數周乃至半年后,尿中仍可檢出其代謝物,且部分代謝物仍具有藥物活性;例如氯丙嗪、異丙嗪等藥物經肝臟代謝,在細胞色素P450酶催化下被氧化為亞砜結構物質,會引起白細胞減少和粒細胞缺乏癥,從而引起人體肝臟、腎臟的病變,還會引起眼部并發癥等[4]。抗組胺類藥物在養殖業中的使用已經引起了美國、歐盟和日本等國家的高度重視。
目前有關動物源性食品中多種抗組胺類藥物的測定方法還較少,鮮見對同時測定動物源性食品中19 種抗組胺類藥物及其代謝物的研究。相關文獻主要僅檢測氯丙嗪、異丙嗪及其代謝物[5-7],大部分研究局限于法醫學方面[8-10]。國內外檢測低濃度水平的獸藥殘留通常采用的方法為液相色譜-串聯質譜法[11-15]和氣相色譜-質譜法[16-17],其中液相色譜-串聯質譜法數據通量大,無需衍生化且定性能力強,是一種靈敏度和選擇性極高的檢測手段。由于動物源性食品種類多、基質復雜,標準方法和文獻報道多數采用固相萃取柱[18-19]、多功能凈化柱[20]等凈化手段,其步驟復雜、實驗成本高。本研究旨在建立一種基于超高效液相色譜-串聯質譜(ultra performance liquid chromatography-tandem mass spectrometry,UPLC-MS/MS),結合空間位阻凈化技術[21-22],實現同時測定19 種抗組胺類藥物及其代謝物殘留。本方法簡便快速,靈敏度滿足實際樣品的檢測需要。
1 材料與方法
1.1 材料與試劑
乙酸乙酯、乙腈、甲酸(均為色譜純) 德國Merck公司;檸檬酸鈉、檸檬酸氫鈉、氯化鈉、硫酸鎂、Na2EDTA(均為優級純) 上海譜振生物科技有限公司;EMR-Lipid吸附劑 美國Agilent公司;實驗用水為Millipore-iQ高純水凈化儀制得。
19 種抗組胺藥物標準品(特非那定、阿司咪唑、西替利嗪、氯雷他定、羥嗪、溴苯那敏、地氯雷他定、賽庚定、氯苯那敏、曲吡那敏、苯海拉明、氯丙嗪、氯丙嗪亞砜、異丙嗪、異丙嗪亞砜、氟奮乃靜、氟奮乃靜亞砜、奮乃靜、奮乃靜亞砜) 德國Dr.Ehrenstorfer公司。
1.2 儀器與設備
1260II超高效液相色譜儀-串聯6470三重四極桿質譜儀 美國Agilent公司;MultiReax旋渦振蕩器 德國Heidolph公司;Centrifuge 5804R高速冷凍離心機 德國Eppendorf公司;N-EVAP112水浴式氮吹儀美國Organomation公司;Millipore-iQ高純水凈化儀美國Millipore公司;AS 3120超聲波振蕩器 天津特賽恩斯儀器有限公司。
1.3 方法
1.3.1 預處理
1.3.1.1 空間位阻凈化法
稱取經均質的試樣5.0 g于50 mL聚丙烯離心管中,加入5 mL超純水、20 mL的乙酸乙酯-乙腈(20∶80,V/V)提取液,勻漿后超聲提取10 min,離心(8 000 r/min,5 min)后取清液加入2 g預先用3 mL水活化的EMR-Lipid吸附劑,渦旋振蕩并離心(8 000 r/min,5 min);取全部清液加入5 g鹽(NaCl-MgSO4,1∶4,m/m),立即劇烈振搖并離心(4 ℃,8 000 r/min,5 min);取上清液在40 ℃水浴中氮吹至盡干,加入1 mL初始比例的流動相(含0.1 mol/L Na2EDTA)復溶后過0.22 μm濾膜供UPLCMS/MS測定。
1.3.1.2 CEN法
稱取經均質的試樣5.0 g于50 mL聚丙烯離心管中,分別加入10 mL超純水、10 mL甲酸-乙腈(10∶90,V/V)提取液,超聲30 min,加入1 g檸檬酸鈉、1 g氯化鈉、4 g硫酸鎂、0.5 g檸檬酸氫鈉,渦旋振蕩并離心(5 000 r/min,5 min)。取5 mL上清液加入0.75 g硫酸鎂、0.25 g N-丙基乙二胺(N-propylethylenediamine,PSA)、0.15 g C18和0.15 g Al-N,渦旋振蕩并離心(5 000 r/min,5 min),取上清液1 mL用0.1%甲酸溶液定容至10 mL,混勻后過0.22 μm濾膜供UPLC-MS/MS測定。
1.3.1.3 AOAC法
稱取經均質的試樣5.0 g于50 mL聚丙烯離心管中,加入10 mL超純水、25 mL乙腈,勻漿提取后,分別加入10 g硫酸鎂、1 g氯化鈉,勻漿并離心(5 000 r/min,5 min)。取上層乙腈4 mL加入1.2 g硫酸鎂、0.1 g PSA、0.1 g C18,渦旋振蕩并離心(5 000 r/min,5 min),取上清液過0.22 μm濾膜供UPLC-MS/MS測定。
1.3.2 標準溶液的配制
標準儲備液(1 mg/mL)的配制:準確稱取19 種藥物標準物質各10.0 mg,用乙腈分別溶解定容至10.0 mL,于-18 ℃冷凍保存。
標準混合儲備液(10 μg/mL)的配制:分別取標準儲備液1.0 mL混合,用乙腈定容至100 mL,于-18 ℃冷凍保存。
標準混合空白工作液的配制:取標準混合儲備液,用初始比例流動相稀釋成1、2、5、10、20、50、100 μg/L系列質量濃度的標準混合空白工作液。
基質匹配混合標準溶液配制:取陰性樣品,按照1.3.1節的預處理方法處理濃縮至近干,加入標準混合空白工作液1.0 mL后按1.3.1節繼續操作,即得。
1.3.3 UPLC-MS/MS測定
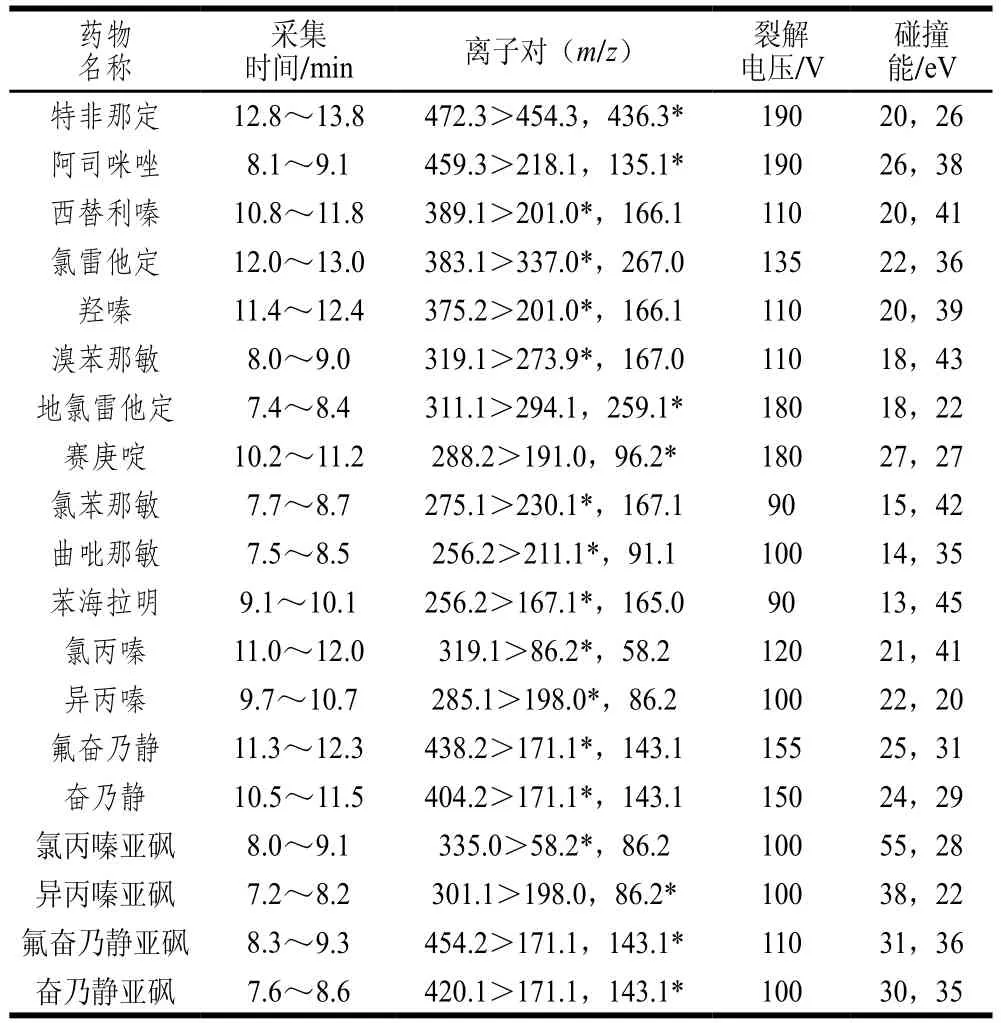
表1 19 種藥物的質譜采集參數Table 1 Mass spectrometric acquisition parameters of 19 drugs
LC條件:Poroshell 120 EC-C18柱(100 mm×2.1 mm,2.7 μm);柱溫35 ℃;流速0.3 mL/min;進樣量10 μL;流動相A為0.1%甲酸溶液,B為0.1%甲酸-乙腈溶液;梯度洗脫程序:0~3 min,10%~10% B;3~6 min,10%~30% B;6~8 min,30%~40% B;8~12 min,40%~70% B;12~14 min,7%~90% B;14~16 min,90%~90% B;16~16.5 min,90%~10% B。
MS條件:Agilent Jet Stream電噴霧離子源;動態多反應監測模式采集數據;干燥氣溫度350 ℃;干燥氣流量10 L/min;鞘氣流量12 L/min;霧化氣壓力25 psi;毛細管電壓4 000 V;MS/MS分辨率為unit;19 種抗組胺藥物的質譜參數見表1。
定量方法:取1.3.2節制備的樣液上機測定繪制標準曲線,再取1.3.1節制備的樣液上機測定,以保留時間和2 對特征離子對定性,以定量離子的峰面積計算樣液中目標物的含量。
2 結果與分析
2.1 質譜條件的優化
在電噴霧離子源正離子全掃描模式下分別對19 種質量濃度為1 μg/mL的抗組胺藥物進行分析,所有待測物均形成[M+H]+形式的準分子離子峰。在單離子掃描模式下分別優化每個待測物的裂解電壓使母離子響應最大,后在子離子掃描和多反應監測分析中確定兩對定性離子對和一對定量離子對,并優化其碰撞能使子離子響應最大化。同時,在多殘留檢測中為盡可能地提升靈敏度和定量的準確性,采用動態多反應監測模式進行數據采集,以根據不同待測物的保留時間,分段進行多反應監測掃描,增加每個待測物的掃描駐留時間,從而加大目標離子的通量提高數據采集效率,進而提升靈敏度、保證重復性。
2.2 色譜條件的優化
因西替利嗪和羥嗪;氟奮乃靜、氟奮乃靜亞砜、奮乃靜和奮乃靜亞砜;氯丙嗪、氯丙嗪亞砜、異丙嗪和異丙嗪亞砜具有結構相同的母核,質譜行為接近,在二級質譜中產生相同的豐度較高的子離子碎片,為準確定量需保證色譜上的分離度。本研究綜合考慮系統耐壓、分析時間和分離效果,主要對比了Poroshell 120 EC-C18柱(100 mm×2.1 mm,2.7 μm)、Eclipse Plus C18柱(100 mm×2.1 mm,1.8 μm)、Cortecs C18柱(100 mm×2.1 mm,2.7 μm)、XBridge C18柱(100 mm×2.1 mm,1.7 μm)和Accucore Vanquish C18柱(100 mm×2.1 mm,1.5 μm)5 種色譜柱。綜合各方面因素最終選取柱效較高且背壓較低的Poroshell 120 C18柱,能達到理想的實驗效果。
根據經驗和文獻資料,酸性流動相體系在正離子模式下能增強待測物的離子化效率從而提高靈敏度,乙腈和甲醇相比具有更小的基線噪音影響和更強的反相洗脫能力,從而增大梯度洗脫程序設計的靈活性,故選擇乙腈為有機相。本研究主要對水相添加不同濃度的甲酸和甲酸銨與乙腈進行組合,考察其響應值,結果以某種水相與A種水相中19 種藥物響應平均值的比值計算,制作變化圖(圖1)。結果表明:水相中加入甲酸和甲酸銨都能明顯提升靈敏度,0.1%的甲酸添加量能達到最佳靈敏度。為保證梯度洗脫的穩定性,同時在乙腈中加入相同的0.1%甲酸。最終確定0.1%甲酸溶液和0.1%甲酸-乙腈溶液作為流動相,所有19 種待測物的典型多反應監測圖見圖2。
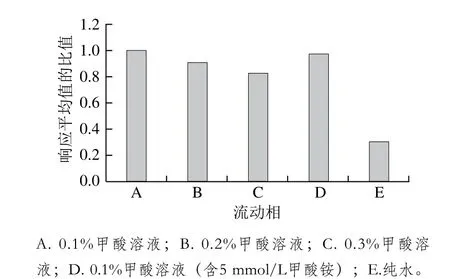
圖1 不同流動相的比較Fig. 1 Comparison of different mobile phases
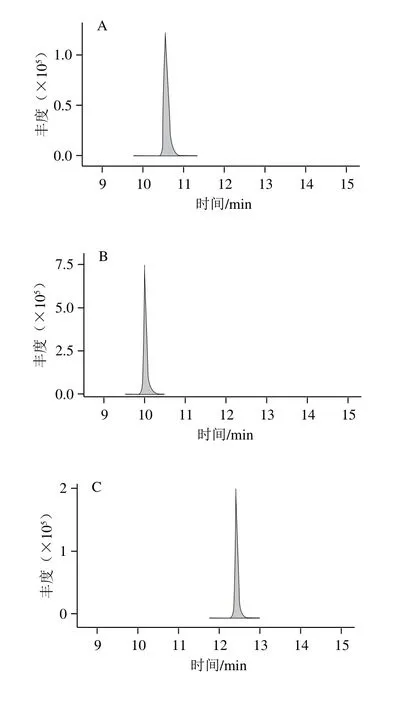
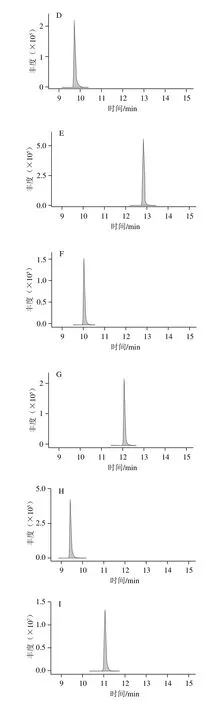
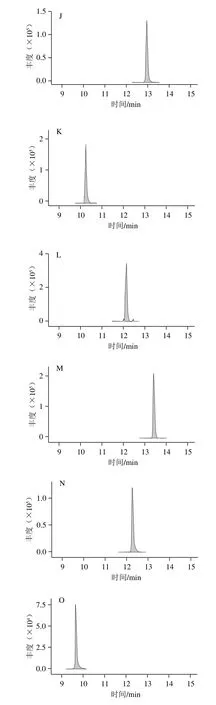

圖2 19 種藥物的典型多反應監測譜圖Fig. 2 Typical MRM chromatograms of 19 drugs
2.3 預處理方法的優化
動物源性食品通常含大量的脂肪、蛋白質等物質,在預處理過程中動物細胞膜的主要成分磷脂也會源源不斷溶出,這些物質在離子源會與待分析物競爭電子引起很大的基質效應,嚴重影響定量的準確性,因此如何最大程度地凈化來削弱基質效應是分析的主要難點。獸藥殘留檢測的常用提取溶劑有緩沖鹽溶液、甲醇、乙腈、乙酸乙酯等,加入適量的甲酸或氨水也能促進提取;19 種抗組胺類藥物極性均較弱但是極性范圍較寬,實驗表明使用緩沖鹽溶液或甲醇作為提取劑時特非那定等藥物的回收率不足40%,使用乙酸乙酯作為提取劑時曲吡那敏等藥物的回收率不足60%,而使用乙腈和乙酸乙酯的混合溶液作為提取劑能覆蓋大部分的待測物的極性;19 種抗組胺類藥物的pKa值范圍在3.7~9.3之間,故不加入甲酸或氨水促進提取,最終選擇乙酸乙酯-乙腈(20∶80,V/V)作為提取溶劑。
分散固相萃取技術是目前實驗室開展藥物殘留檢測工作主流的預處理手段[23-31],它兼顧了凈化效果和效率,AOAC法和CEN法等國際方法中主要用的吸附劑有PSA、石墨化碳黑(graphitized carbon black,GCB)、十八烷基鍵合硅膠(C18)、中性氧化鋁(Al-N)等,其中PSA和Al-N主要吸附極性雜質、GCB主要吸附色素同時會對平面結構的化合物存在共吸附、C18主要吸附脂類和糖類物質但是因針對性較差也引起一些親脂類化合物回收率偏低;EMR-Lipid技術是一種基于空間位阻原理的特殊聚合物基質EMR的凈化手段,專門吸附脂質中C5及以上的碳鏈,對脂質具有非常強的選擇吸附性。由于19 種抗組胺類藥物的極性均較弱,因此在反相色譜上的共餾出物于離子源處形成基質效應的物質主要是磷脂等弱極性的物質,本研究考察了通過空間位阻凈化法、CEN法和AOAC法凈化豬肉樣品后的磷脂殘留來評價3 種預處理方式的凈化效果,磷脂并非單一化合物,主要分為甘油磷脂和鞘磷脂兩大類,但是其含磷酸的母體是相同的,故可采用母離子掃描模式監測184+碎片離子來監測磷脂殘留量。如圖3所示,可知空間位阻凈化法的磷脂殘留最少,CEN法凈化后仍有一定量的磷脂存在,AOAC法凈化后磷脂殘留最多,因此最終確定了空間位阻凈化法為本研究的預處理手段。
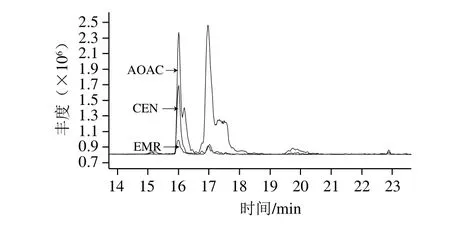
圖3 豬肉樣品經3 種方法凈化后的磷脂殘留監測對比圖Fig. 3 Comparison of phospholipid residues in pork samples purified by three methods
由于氯丙嗪、異丙嗪、奮乃靜、氟奮乃靜等吩噻嗪類抗組胺藥物具有相同的硫氮雜蒽母核,其環上硫原子所具有的兩對孤對電子極易與基質中的金屬離子絡合甚至被儀器金屬管路吸附,造成峰形拖尾、重復性差等現象,Na2EDTA能競爭絡合基質中的金屬離子使待測物母核游離出來,因此最終復溶時用流動相加入Na2EDTA(最終濃度為0.1 mol/L)較好地改善了重復性。
2.4 基質效應的考察結果
液相色譜-質譜分析中存在的基質效應,除了在預處理過程中盡可能凈化樣品,通常采用稀釋法、標準加入法、內標法、基質配標法來削弱或修正基質效應造成的偏差;本研究以基質效應最明顯的豬肝為例,通過優化梯度洗脫程序和基質配標法較好地減弱和抵消了基質效應,比較結果見表2。
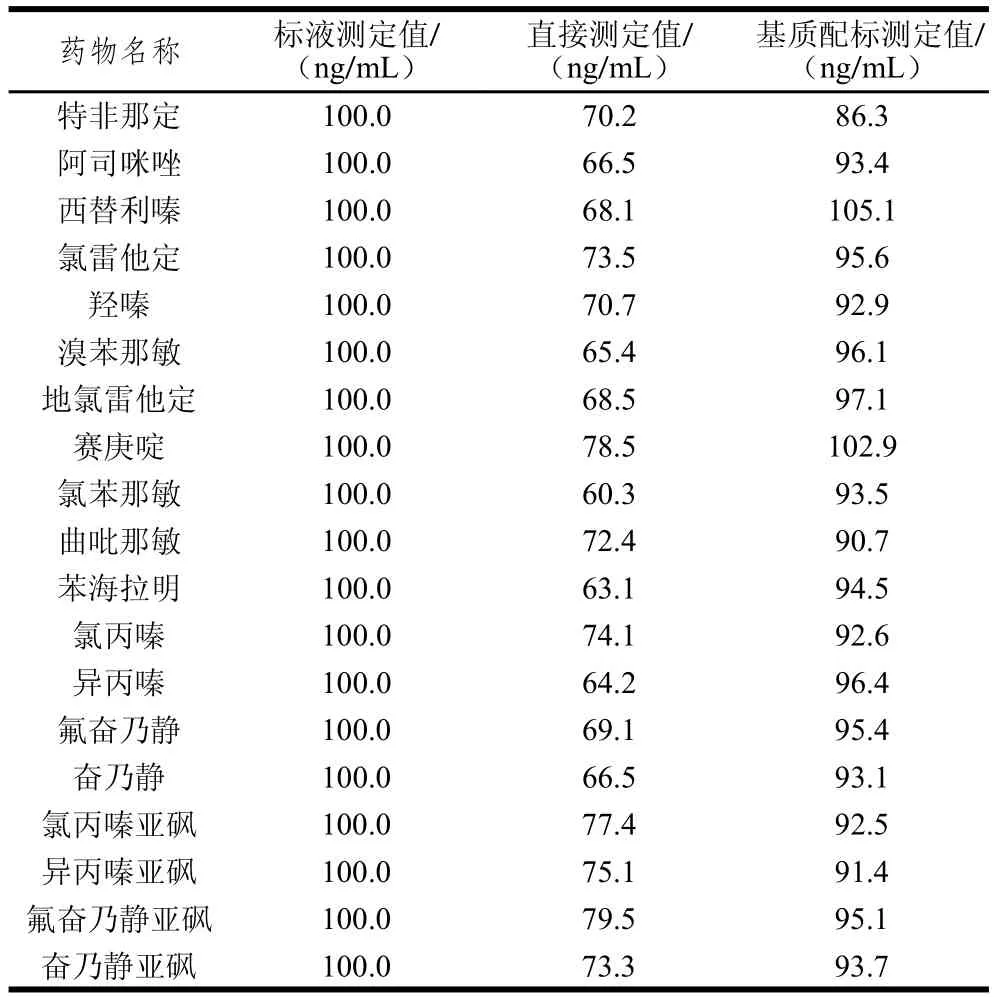
表2 19 種藥物的基質效應Table 2 Matrix effects of 19 drugs
2.5 方法學考察結果
2.5.1 線性與檢出限結果
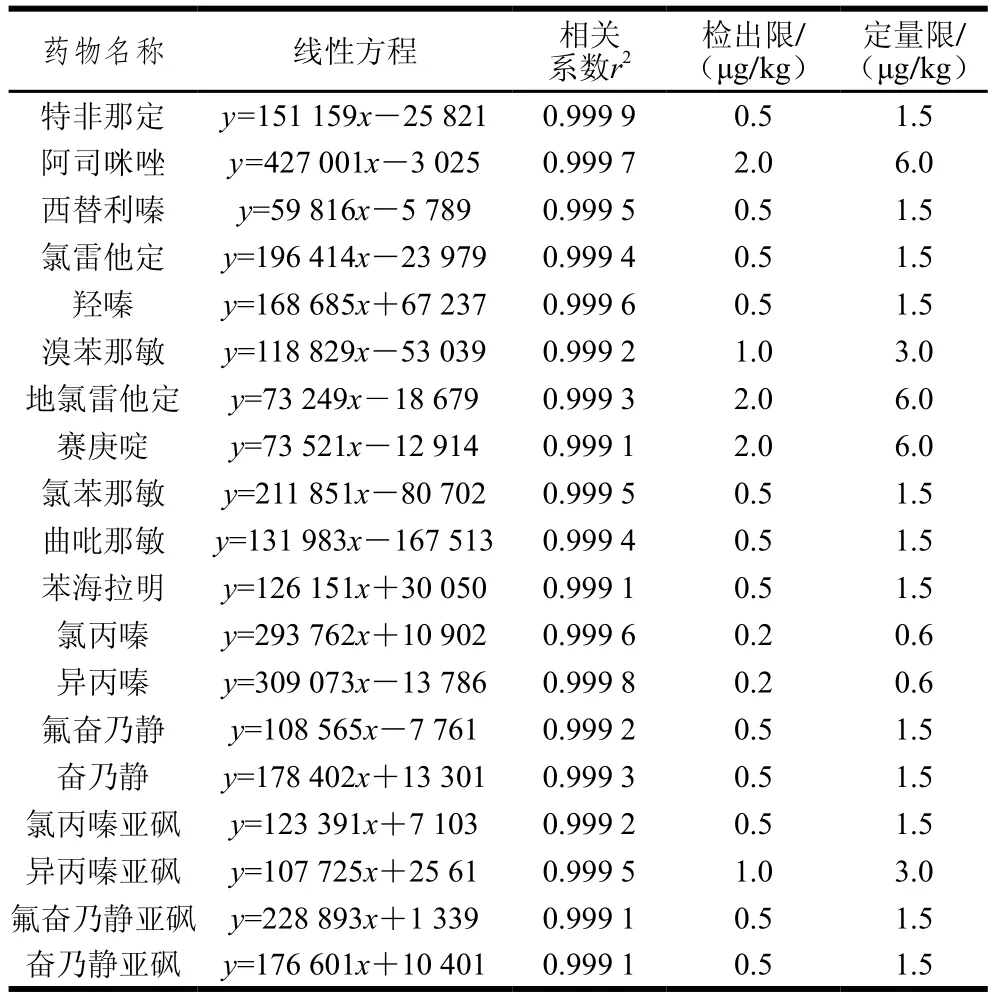
表3 19 種藥物的線性范圍、線性方程、相關系數、檢出限和定量限Table 3 Linear ranges, regression equations, correlation coefficients, LODs and LOQs of 19 drugs
按上述條件將系列混合標準工作溶液上機測定,以峰面積為縱坐標(y),以質量濃度(ng/mL)為橫坐標(x)制作標準工作曲線,得到線性方程的線性回歸系數r2均大于0.999,表明19 種抗組胺藥物在相應的質量濃度范圍內線性良好。采用標準添加法進行測定,以定性子離子的3 倍信噪比計算檢出限和10 倍信噪比計算定量限,結果見表3,得到19 種抗組胺藥物檢出限為0.2~2.0 μg/kg,定量限為0.6~6.0 μg/kg,靈敏度滿足實際樣品檢測的需要。
2.5.2 回收率與重復性結果
在陰性牛肉、雞肉和豬肝中分別添加3個質量濃度水平的混合標準溶液進行6 次回收率實驗,結果見表4。可知回收率在75.1%~89.1%之間,相對標準偏差為2.3%~7.1%,方法具有較好的精密度,能滿足實際樣品的檢測需要。
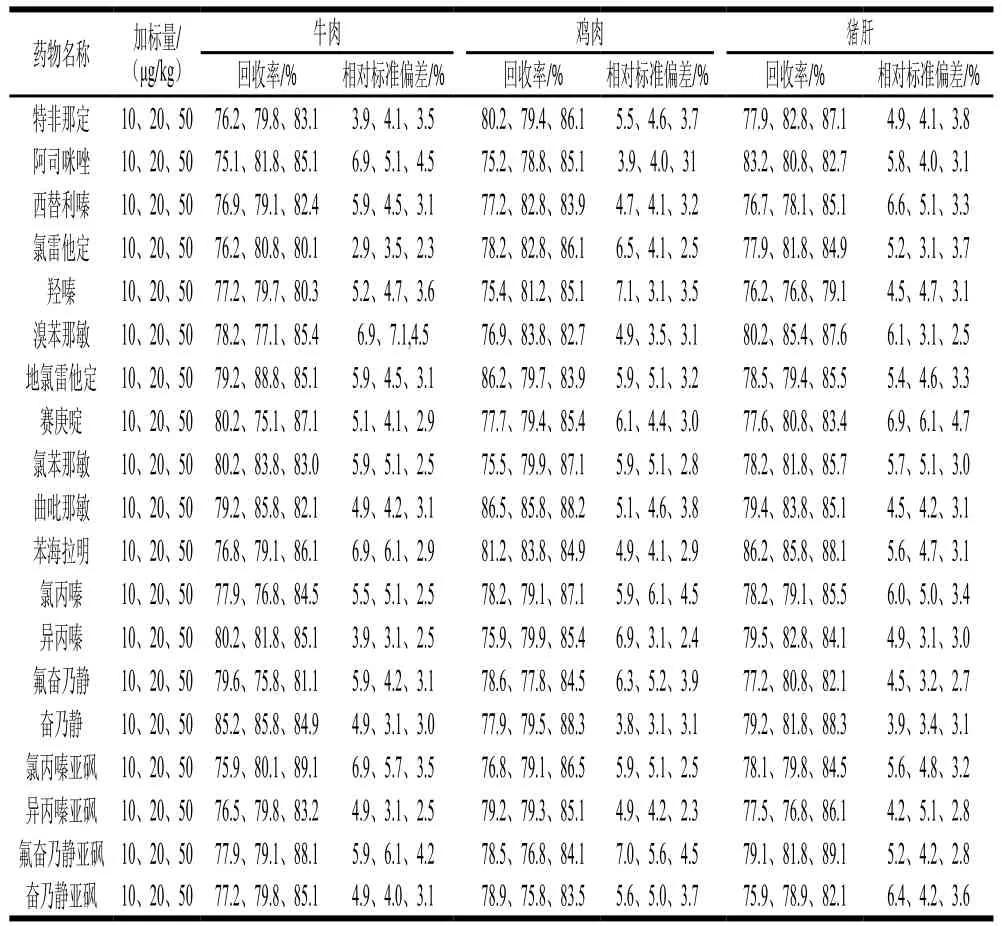
表4 19 種獸藥的準確度和精密度(n=6)Table 4 Accuracy and precision for 19 drugs (n= 6)
2.5.3 實際樣品檢測結果
采用本方法對流通領域市售雞肉86 份、牛肉91 份、豬肝77 份進行19 種抗組胺藥物及其代謝物的篩查,檢出豬肝氯丙嗪陽性樣品3 例、異丙嗪陽性樣品1 例,檢出牛肉氯丙嗪陽性樣品1 例、奮乃靜陽性樣品1 例;全部陽性樣品同時均檢出其亞砜結構代謝物,可見通常代謝物與本體會同時存在,內臟等脂肪含量高的組織更易蓄積抗組胺類藥物及其代謝物。
3 結 論
本實驗建立UPLC-MS/MS同時測定畜禽肉中19 種抗組胺藥物及其代謝物殘留的方法。通過比較空間位阻凈化法、CEN法和AOAC法3 種不同前處理方法的凈化效果,驗證了空間位阻凈化法的有效性。通過優化色譜和質譜參數,得到了最佳的儀器檢測條件。方法學考察和實際樣品的測定證明該方法實用、可靠。與目前的國家標準和文獻報道的方法相比,本方法在擴大了同類型藥物檢測范圍、準確性較好、靈敏度較高的同時,盡可能省去了復雜的前處理,節省了成本,能更快更好地為市場監管部門的監督執法提供技術支持。
