CoMo/MgO催化劑的制備及其對4,6-二甲基二苯并噻吩的加氫脫硫性能
2019-08-01 01:48:10鄭世富何明陽
石油學報(石油加工) 2019年4期
鄭世富,張 磊,何明陽
(常州大學 石油化工學院,江蘇 常州 213164)
隨著社會的發展,人們對環境的要求日益提高。世界各國環保部門對燃油中的氮、硫等含量都有了更高標準:歐洲地區于2013年實施歐Ⅶ汽車排放標準(汽、柴油硫質量分數<10 μg/g);北美洲以美國為首的發達國家,在2010年全面推廣硫質量分數小于10 μg/g的柴油,并延續至今[1-2]。我國現行油品標準中,柴油中硫含量要高于發達國家,2019年我國實施車用柴油國Ⅵ標準,全面供應硫質量分數不大于10 μg/g的普通柴油[3]。近年來,隨著中國經濟的迅速發展,對原油的需求量正在逐漸增長;同時隨著世界范圍內石油資源的日益減少,重質化原油比例加大。因此,在油品加氫精制領域,清潔燃油的生產面臨著嚴峻的挑戰。
加氫脫硫反應中的催化劑載體一直是研究熱點,氧化鋁(γ-Al2O3)是加氫脫硫傳統催化劑載體,由于其高的比表面積和良好的機械性能,因此成為加氫精制催化劑的首選載體。但是,傳統γ-Al2O3負載的金屬硫化物催化劑對油品中最難脫除的4,6-二甲基二苯并噻吩(4,6-DMDBT)的催化活性較低,在現有工藝條件下實現對其深度脫除是有困難的。要得到超低硫含量的清潔油品,需要較高的反應溫度和壓力,因而增加了操作成本[4]。因此,人們嘗試使用其他載體來改善金屬硫化物催化劑的催化性能,如:混合氧化物[5]、分子篩[6-8]、活性炭[9-10]等。但是,混合氧化物、分子篩等多為酸性載體,導致反應過程中催化劑表面易積炭,而降低催化活性[11]。
氧化鎂(MgO)作為堿性氧化物,廣泛應用于催化領域中,如:水煤氣變換[12]、丙烯環氧化[13]、加氫精制[14]等。由于MgO載體表面存在堿中心,在催化劑浸漬過程中,酸性的氧化鉬物種與MgO表面的堿中心在一定程度上發生酸-堿作用,使得相對顆粒尺寸較小的氧化鉬物種分散在MgO表面,經過還原硫化后形成分散度較高的硫化物活性相。因此,MgO負載的金屬硫化物催化劑,具有較高的加氫脫硫活性[14-16]。筆者所在課題組前期工作中,利用加壓碳化的方法制得了高比表面積的氧化鎂納米片(MgO),并負載CoMo催化劑,在二苯并噻吩的加氫脫硫反應中具有很好的催化性能[17]。作為持續的工作,本文中筆者采用高比表面積的MgO為載體,使用四硫代鉬酸銨作為Mo的前軀體,制備負載型CoMo催化劑(CoMo/MgO-S),考察催化劑對油品中難以脫除的4,6-DMDBT的催化性能。進一步以鉬酸銨為Mo的前軀體,獲得 CoMo/MgO-O 催化劑,并研究不同催化劑上Mo物種的硫化程度和金屬活性相的分散狀態。
1 實驗部分
1.1 試劑和原料
六水合硝酸鈷(Co(NO3)2·6H2O)和四水合鉬酸銨((NH4)6Mo7O24·4H2O),分析純,國藥集團化學試劑有限公司產品;4,6-二甲基二苯并噻吩(4,6-DMDBT,質量分數99%),紐安節化工科技有限公司產品;硫化銨((NH4)2S)水溶液(質量分數20%),阿拉丁公司產品;十氫萘(質量分數98%),成都聯合化工有限公司產品;無水乙醇,分析純,江蘇永豐化學試劑有限公司產品;氧化鎂(MgO),自制,具體制備過程請參考文獻[17];氧化鋁(γ-Al2O3),工業級,上海恒業分子篩股份有限公司產品。
1.2 四硫代鉬酸銨的制備
在100 mL圓底燒瓶中加入2.665 g (NH4)6Mo7O24·4H2O,然后再加入20 mL蒸餾水,攪拌使其充分溶解,在60 ℃油浴中預熱20 min,最后加入35.6 mL的(NH4)2S水溶液,在60 ℃油浴處理1 h后,將所得溶液于冰箱中冷卻結晶,抽濾,并用無水乙醇洗滌,室溫下晾干,得到四硫代鉬酸銨((NH4)2MoS4)前軀體。
1.3 催化劑的制備
CoMo/MgO-S催化劑的制備采用分步浸漬法:取1 g MgO載體,將含有0.2903 g (NH4)2MoS4的水溶液浸漬到MgO載體上,在100 ℃下干燥 12 h 后,等體積浸漬Co(NO3)2·6H2O溶液。所得樣品在室溫下放置12 h,100 ℃下干燥12 h。然后將干燥后的樣品在10 MPa下壓片成型、壓碎過篩,得到粒徑為250~380 μm的顆粒。
CoMo/MgO-O催化劑的制備采用等體積浸漬法:取1 g MgO載體,稱取0.1982 g的(NH4)6Mo7O24·4H2O與0.1618 g的Co(NO3)2·6H2O,其中Co與Mo的摩爾比為1∶2,并溶于一定量的蒸餾水中,然后將所得溶液等體積浸漬到MgO載體上。樣品在室溫下放置12 h,100 ℃下干燥12 h,然后將干燥后的樣品在10 MPa下壓片成型、壓碎過篩,得到粒徑為250~380 μm的顆粒。
作為比較,以傳統γ-Al2O3為載體,采用相同的方法負載CoMo催化劑,標記為CoMo/γ-Al2O3-O。
上述催化劑均在H2-H2S(15%體積分數H2S)混合氣體中400 ℃硫化3 h,升溫速率為2 ℃/min,氣體流量為30 mL/min。其中,Co和Mo的負載質量分數均分別為3.3%和10.7%。
1.4 催化劑的表征
樣品的X-射線多晶粉末衍射(XRD)分析在RIGAKU Smart Lab衍射儀上完成,Cu靶Kα輻射,管電壓40 kV,管電流100 mA,2θ掃描范圍5°~80°。
氮氣的吸附-脫附曲線在Micromeritics ASAP 2020M設備上獲得。將樣品于200 ℃下脫氣處理8 h,樣品的表面積和孔徑分布分別采用Brunner-Emmet-Teller(BET)方法和Barrett-Joyner-Halenda(BJH)方法計算。
X-射線光電子能譜分析(XPS)在ESCALAB MK II設備上完成。
樣品的透射電鏡(TEM)照片在JEM-2100型設備上獲得。將硫化后的樣品于無水乙醇中超聲分散0.5 h,然后用毛細管滴加在碳膜包覆的銅網上。通過大量的TEM照片,對MoS2活性相的堆垛層數和長度進行統計分析。假定MoS2活性相是規則的六邊形,估算MoS2分散度(DMo)[18-19]。DMo定義為位于MoS2邊緣表層Mo原子數與總的Mo原子數的比值,可以采用公式(1)和(2)進行計算[20-22]:
(1)
Li=0.32(2ni-1)
(2)
式中,ni為MoS2相中邊緣Mo原子的數量;t是統計TEM圖片中MoS2條紋數目;Li為所統計的第i條MoS2顆粒長度,nm。
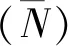
(3)
(4)
式中,Ni是所統計的第i條MoS2顆粒堆垛層數;yi是所統計的第i條MoS2顆粒堆積層數為Ni的數目;xi是所統計的第i條MoS2顆粒長度為Li的數目。
1.5 催化劑活性評價
催化劑的活性測試在固定床反應裝置上進行,反應器內徑為6 mm。硫化后的催化劑取0.3 g,采用石英砂作為稀釋劑,4,6-DMDBT為模型化合物,十氫萘為溶劑,其中4,6-DMDBT的質量分數(w0)為0.47%。反應條件為:反應溫度300 ℃、氫分壓5.0 MPa、氫/油體積比715、質量空速(MHSV)45 h-1。每隔1 h采集1次,使用配備FID檢測器的Agilent 7890B GC分析。
在消除質量傳遞和熱量傳遞條件下,比較催化劑的本征加氫脫硫活性。實驗采用0.08 g硫化的催化劑,與石英砂充分混合后裝入反應器。反應條件為:溫度300 ℃、氫分壓5.0 MPa、氫氣流量50 mL/min、4,6-DMDBT的質量分數0.47%、MHSV范圍35~70 h-1。
4,6-DMDBT加氫脫硫的反應速率常數(kHDS)和轉化頻率(TOF)由公式(5)和(6)進行計算[18]:
(5)
(6)
式中,x是4,6-DMDBT的轉化率,%;F是4,6-DMDBT 的摩爾流量,mol/s;NMo是催化劑中Mo原子的物質的量,mol;m是催化劑的質量,g。
2 結果與討論
2.1 合成的不同Mo源負載的CoMo/MgO-S和CoMo/MgO-O催化劑的物性表征
2.1.1 XRD表征
圖1為所合成的(NH4)2MoS4、MgO載體、CoMo/MgO-S和CoMo/MgO-O的XRD圖譜。圖1(a)中衍射峰的位置與(NH4)2MoS4的標準卡片(PDF#48-1662)中特征衍射峰的位置完全一致,說明合成的晶體材料為(NH4)2MoS4。由圖1(b)可見,2θ=42.7°和61.8°處為所用MgO載體的特征衍射峰。在經過硫化的催化劑樣品CoMo/MgO-S和CoMo/MgO-O催化劑上,均沒有檢測到與Co和Mo物種相關的特征衍射峰,表明硫化物活性相很好地分散在催化劑上。
2.1.2 N2吸附-脫附表征
圖2給出了MgO載體及其使用不同Mo源負載CoMo催化劑的N2吸附-脫附等溫線和孔徑分布圖。由圖2(a)可知,MgO載體的N2吸脫附曲線在相對壓力0.45~0.95區間存在明顯的滯后環,說明MgO載體具有介孔結構,對應的孔徑分布圖顯示其孔徑大小分別集中在3.3和10.0 nm附近。同時吸脫附曲線呈現出V型特征,表明載體材料為狹縫介孔型結構。類似地,CoMo/MgO-S和CoMo/MgO-O催化劑N2吸附-脫附曲線也存在明顯滯后環,但是,CoMo/MgO-O滯后環明顯地向著高比壓區偏移。因此,2個催化劑呈現了不同的孔徑分布,CoMo/MgO-S 催化劑的孔徑大小主要集中在3.6 nm,而CoMo/MgO-O的主要集中在15.1 nm。表1列出了MgO載體及CoMo/MgO-S和 CoMo/MgO-O 催化劑的比表面積、孔體積和平均孔徑大小。由表1可知,與MgO載體相比,以不同Mo源制備催化劑并經硫化處理后,樣品的比表面積均有所降低,表明CoMo催化劑沉積到MgO載體表面或孔結構中。其中,催化劑CoMo/MgO-S的介孔體積有所減小,而CoMo/MgO-O的介孔孔體積增加。這可能與催化劑制備過程中浸漬液中的水溶劑與MgO反應有關。進一步比較發現,以四硫代鉬酸銨為前軀體制備的CoMo/MgO-S催化劑比表面積為161 m2/g,要高于以鉬酸銨采用等體積浸漬制備的CoMo/MgO-O催化劑(129 m2/g)。比表面積較大有利于增加催化劑與反應物的接觸,提高催化劑的HDS活性。
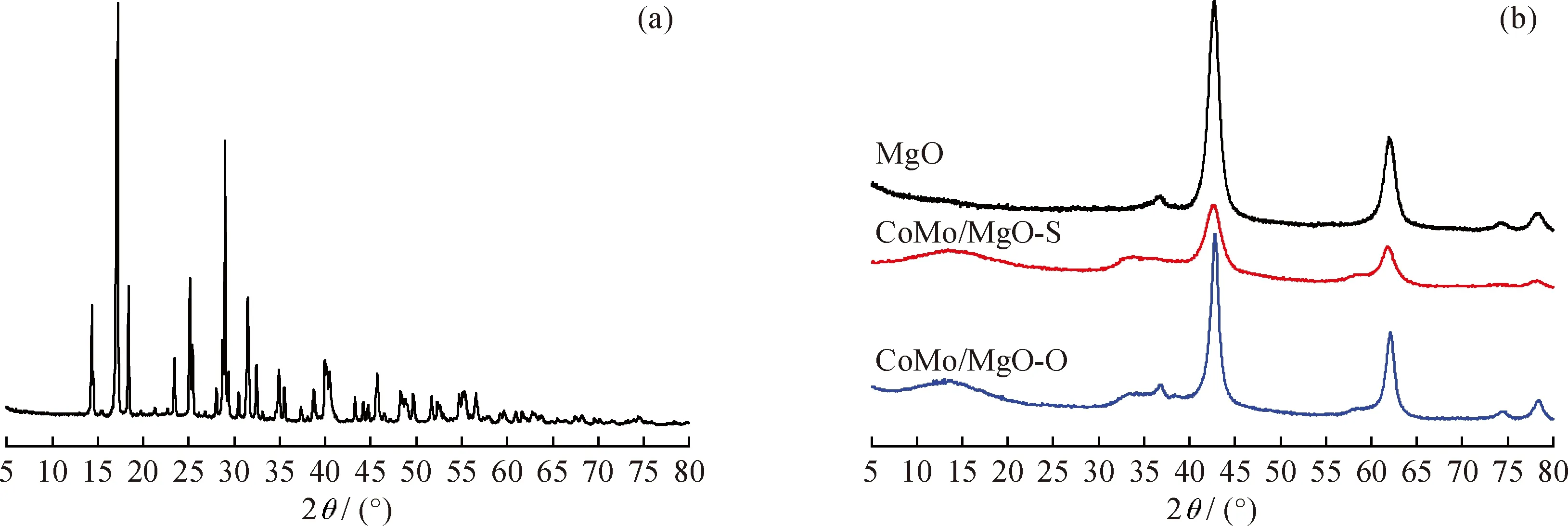
圖1 (NH4)2MoS4、MgO載體及CoMo/MgO-S、CoMo/MgO-O催化劑的XRD圖譜Fig.1 XRD patterns of (NH4)2MoS4,MgO support,CoMo/MgO-S and CoMo/MgO-O catalysts(a)(NH4)2MoS4;(b)MgO,CoMo/MgO-S and CoMo/MgO-O
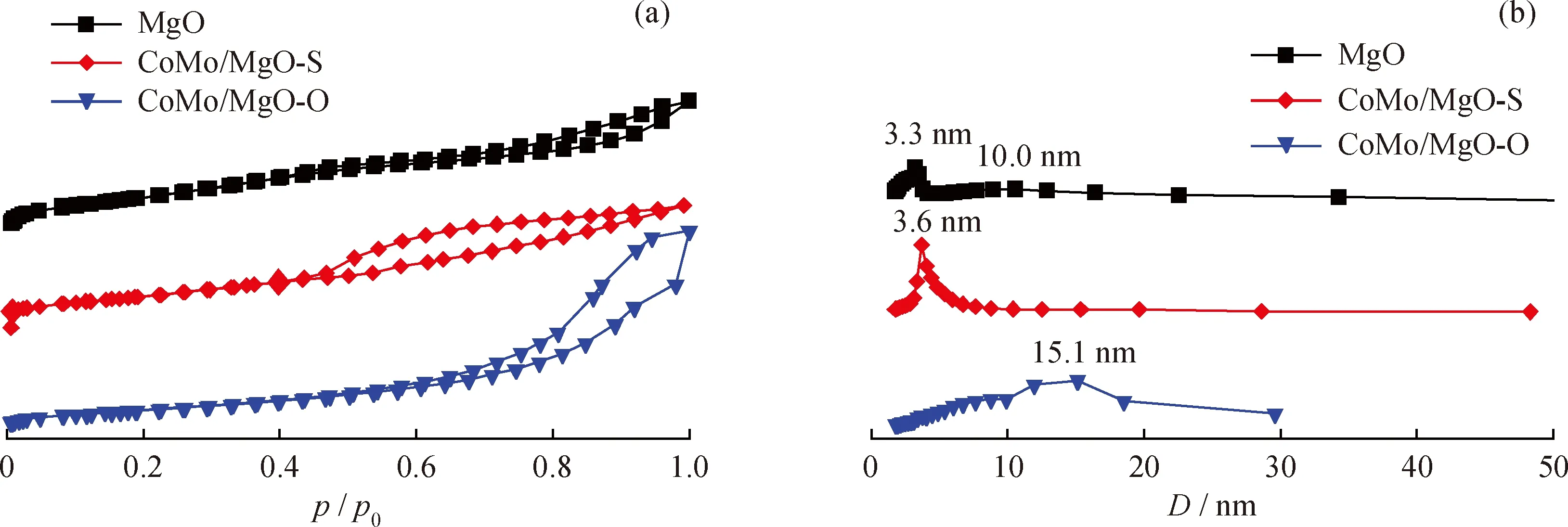
圖2 MgO載體及CoMo/MgO-S和CoMo/MgO-O催化劑的N2吸附-脫附等溫線和孔徑分布Fig.2 Nitrogen adsorption-desorption isotherms and pore size distribution of MgO support,CoMo/MgO-S and CoMo/MgO-O catalysts(a)N2 adsorption-desorption isotherm;(b)Pore size distribution

表1 MgO載體及CoMo/MgO-S和CoMo/MgO-O催化劑的織構參數Table 1 Textural parameters of MgO support, CoMo/MgO-S and CoMo/MgO-O catalysts
1)SBET—BET surface area;2)Vmeso—Mesoporous volume;3)D—Average pore diameter
2.1.3 XPS表征
為了研究硫化后催化劑表面Co和Mo物種的價態與分布情況,對催化劑進行了XPS分析,其結果見圖3。根據文獻[17]報道,對于金屬硫化物催化劑,表面Mo物種以MoS2、MoOxSy和MoO33種形式存在,對應的電子結合能分別是(228.6±0.3)eV (MoS2,Mo4+)、(230.5±0.2)eV (MoOxSy,Mo5+)和(231.2±0.2)eV (MoO3,Mo6+)。其中,Mo4+的含量代表硫化物催化劑的硫化程度,與催化劑的加氫脫硫性能密切相關。一般地,硫化程度越高,催化劑的加氫脫硫性能越好。因此,對CoMo/MgO-S 和CoMo/MgO-O催化劑上Mo物種的XPS光譜(圖3(a))進行分峰擬合分析,并計算2個催化劑上不同Mo物種的相對含量,結果見表2。由表2可知,CoMo/MgO-S催化劑上Mo物種的硫化程度為86.0%,高于CoMo/MgO-O催化劑(79.6%),說明有更多的Mo物種轉化成MoS2活性相。這是因為(NH4)2MoS4前軀體中Mo-S的相互作用弱于(NH4)6Mo7O24前軀體中的Mo—O鍵,在催化劑硫化過程中有利于Mo物種還原硫化成MoS2活性相[26]。同時,2個催化劑上均存在Co-(II)、CoMoS和Co9S83種Co物種的相,分別對應的電子結合能(781.1±0.5 eV)(Co-(II))、(778.8±0.3 eV)(CoMoS)和(777.9±0.4 eV)(Co9S8)[24]。表3給出了不同Co物種在2個催化劑表面的含量。由表3可見,CoMo/MgO-S催化劑上Co物種參與形成CoMoS活性相的比例為63.8%,多于CoMo/MgO-O催化劑(55.4%),說明在催化劑CoMo/MgO-S上形成了更多的CoMoS活性相。
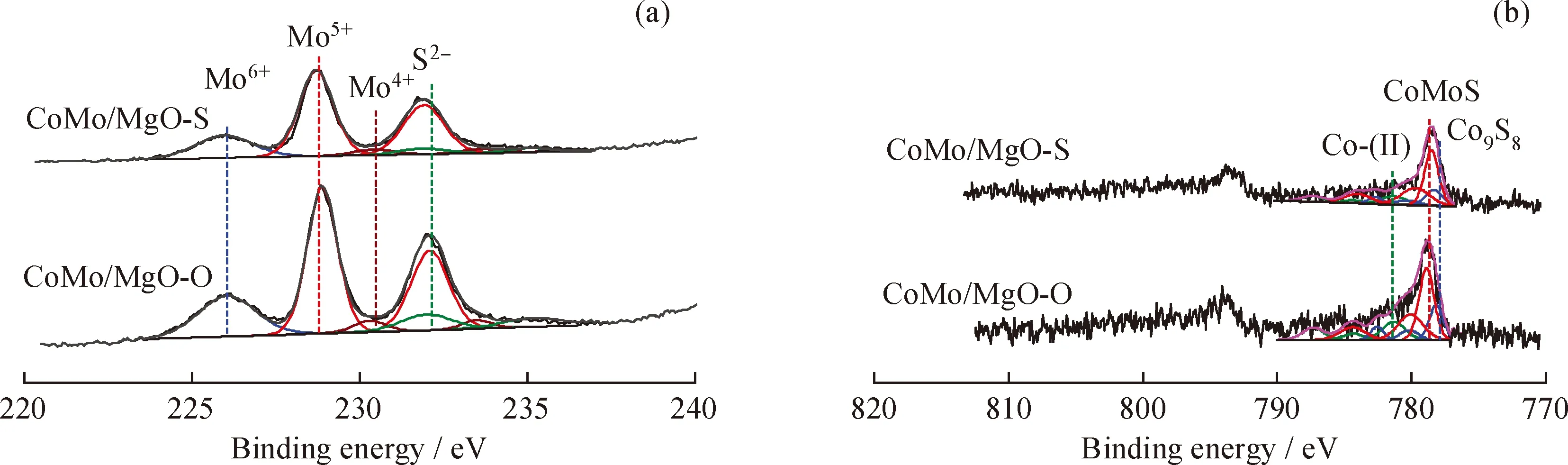
圖3 硫化催化劑CoMo/MgO-S和CoMo/MgO-O的XPS Mo3d和Co2p譜圖及其對應分解峰Fig.3 XPS spectra and decomposition peaks of Mo3d and Co2p of CoMo/MgO-S and CoMo/MgO-O sulfided catalysts(a)Mo3d;(b)Co2p
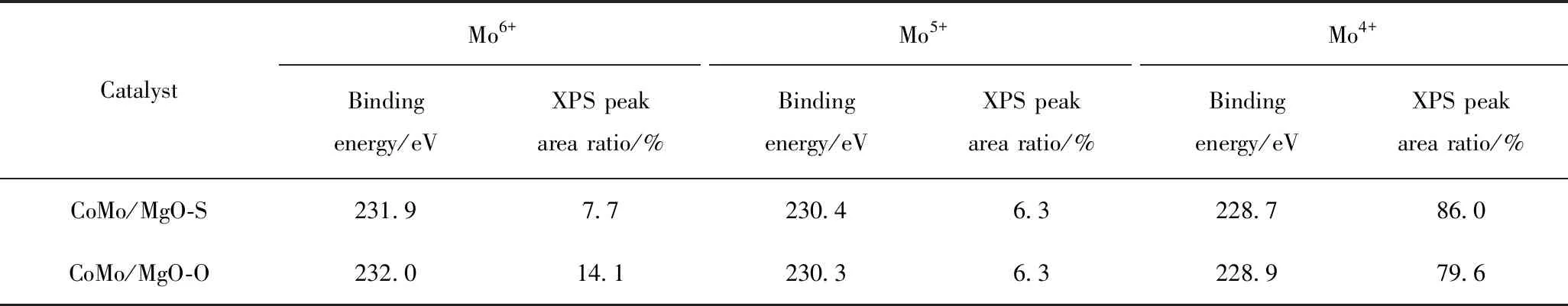
表2 硫化催化劑CoMo/MgO-S和CoMo/MgO-O表面Mo3d物相XPS擬合結果Table 2 Mo3d XPS fitting results of CoMo/MgO-S and CoMo/MgO-O sulfided catalysts

表3 硫化催化劑CoMo/MgO-S和CoMo/MgO-O表面Co2p物相XPS擬合結果Table 3 Co2p XPS fitting results of CoMo/MgO-S and CoMo/MgO-O sulfided catalysts
2.1.4 TEM表征
圖4給出了硫化后CoMo/MgO-S和CoMo/MgO-O催化劑上的TEM照片。由圖4可見,在2個催化劑上均能夠明顯地觀察到MoS2相的晶格條紋,晶面間距約為0.62 nm左右。在這2個催化劑上的不同樣品區域拍攝TEM照片,對每個催化劑上MoS2相的片層長度和堆垛層數進行統計分析,其結果見圖5。可以發現,MoS2相的片層長度在2個催化劑上的分布情況類似,均主要集中在3~6 nm。但是,MoS2相在CoMo/MgO-S上的堆垛層數主要集中在1~3層,甚至可形成4層的MoS2相;而在 CoMo/MgO-O 催化劑上,其堆垛層數主要集中在 1~2 層。表4給出了2個催化劑上MoS2相的平均片層長度、平均堆垛層數和分散度。表4結果也說明了2個催化劑上具有相近的MoS2片層長度,但是CoMo/MgO-S催化劑的平均堆垛層數是2.1,大于CoMo/MgO-O催化劑的。這說明以(NH4)2MoS4為前軀體,更容易形成多層的MoS2相。根據文獻[25-26]報道,MoS2相的加氫性能與其堆垛層數有關,單層的MoS2相(Type I)加氫活性較低,而兩層以上的多層MoS2相(Type II)具有較高的加氫活性。因此可以推測,與CoMo/MgO-O相比,CoMo/MgO-S催化劑應該具有更好的加氫性能。而燃油中的大分子4,6-DMDBT等主要通過加氫脫硫的路徑脫除,改善硫化物催化劑的加氫性能有利于含硫大分子的有效脫硫。
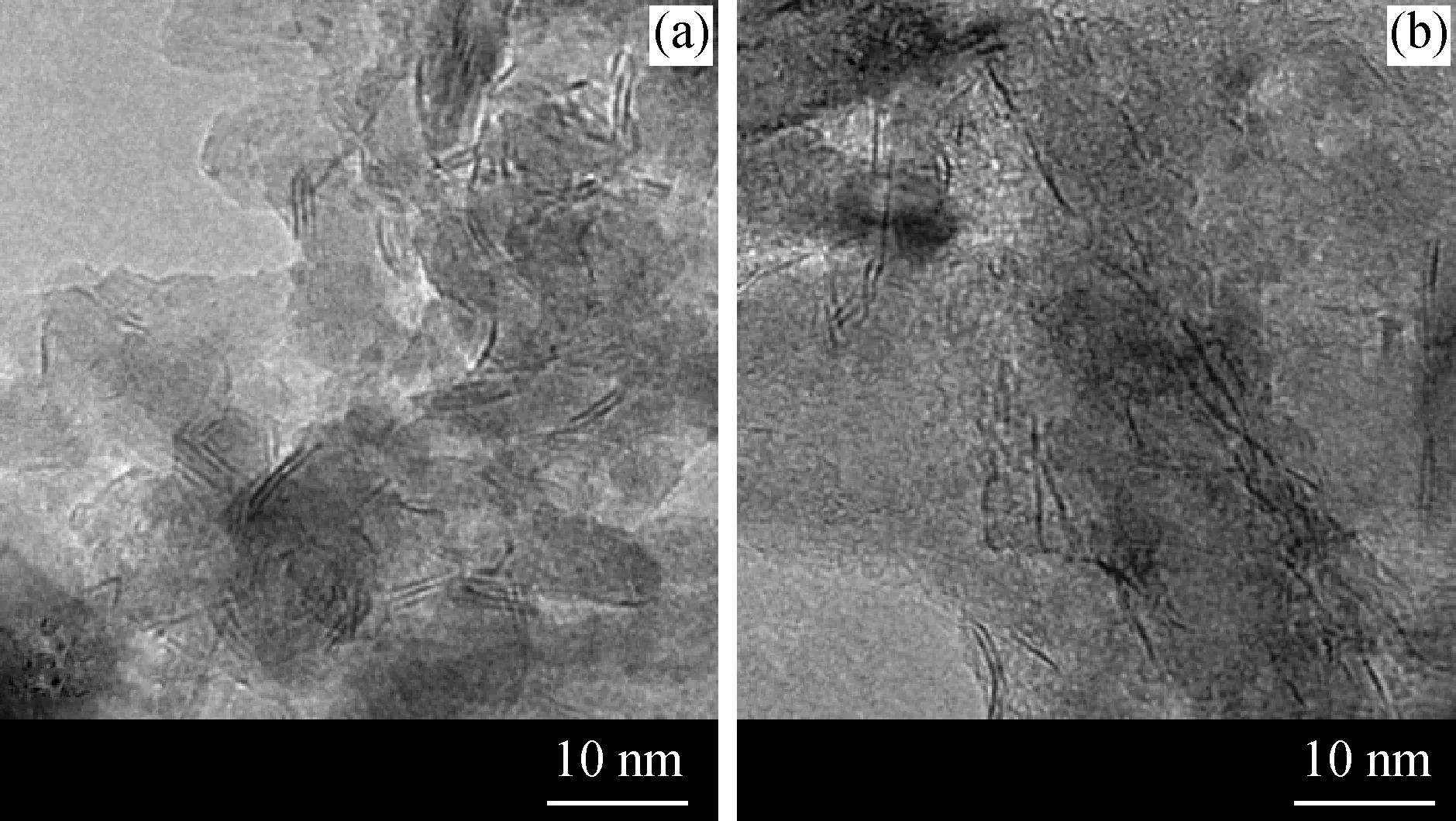
圖4 硫化后CoMo/MgO-S和CoMo/MgO-O催化劑的TEM照片Fig.4 TEM micrographs of CoMo/MgO-S and CoMo/MgO-O sulfided catalysts(a)CoMo/MgO-S;(b)CoMo/MgO-O
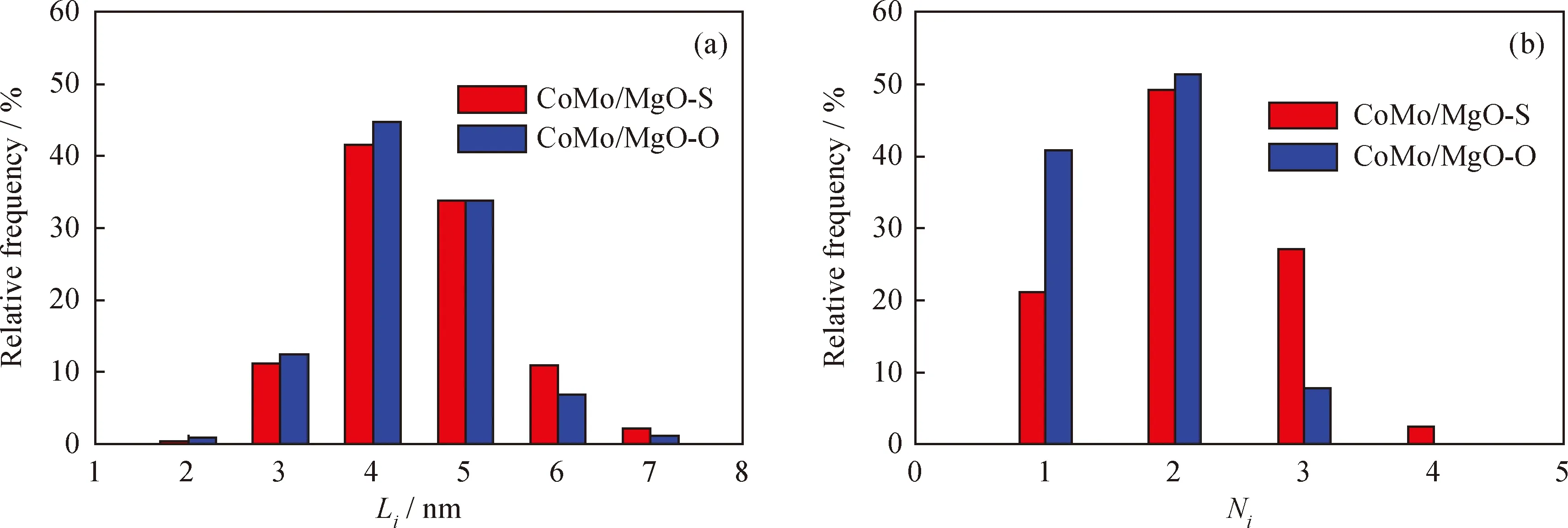
圖5 硫化催化劑CoMo/MgO-S和CoMo/MgO-O的MoS2條紋的長度(Li)、堆垛層數(Ni)分布圖Fig.5 Length (Li)and layer stacking (Ni)distribution of MoS2 slabs of CoMo/MgO-S and CoMo/MgO-O sulfided catalysts(a)Li;(b)Ni
表4 硫化催化劑CoMo/MgO-S和CoMo/MgO-O的MoS2晶體平均長度平均堆垛層數和分散度(DMo)Table 4 Average length number of the stacking layers and dispersity (DMo)of MoS2 crystallites for CoMo/MgO-S and CoMo/MgO-O sulfided catalysts
2.2 CoMo/MgO-S和CoMo/MgO-O催化劑加氫脫硫活性評價結果
以 4,6-DMDBT 為模型化合物,考察了CoMo/MgO-S 和CoMo/MgO-O催化劑的加氫脫硫活性,結果見圖6。由圖6可見:在4,6-DMDBT加氫脫硫反應中,2個催化劑均呈現較好的穩定性;CoMo/MgO-S催化劑的催化性能明顯優于 CoMo/MgO-O 催化劑;反應進行20 h時,CoMo/MgO-S 催化劑上4,6-DMDBT的轉化率為70.8%,而CoMo/MgO-O催化劑上的轉化率僅為52.3%。
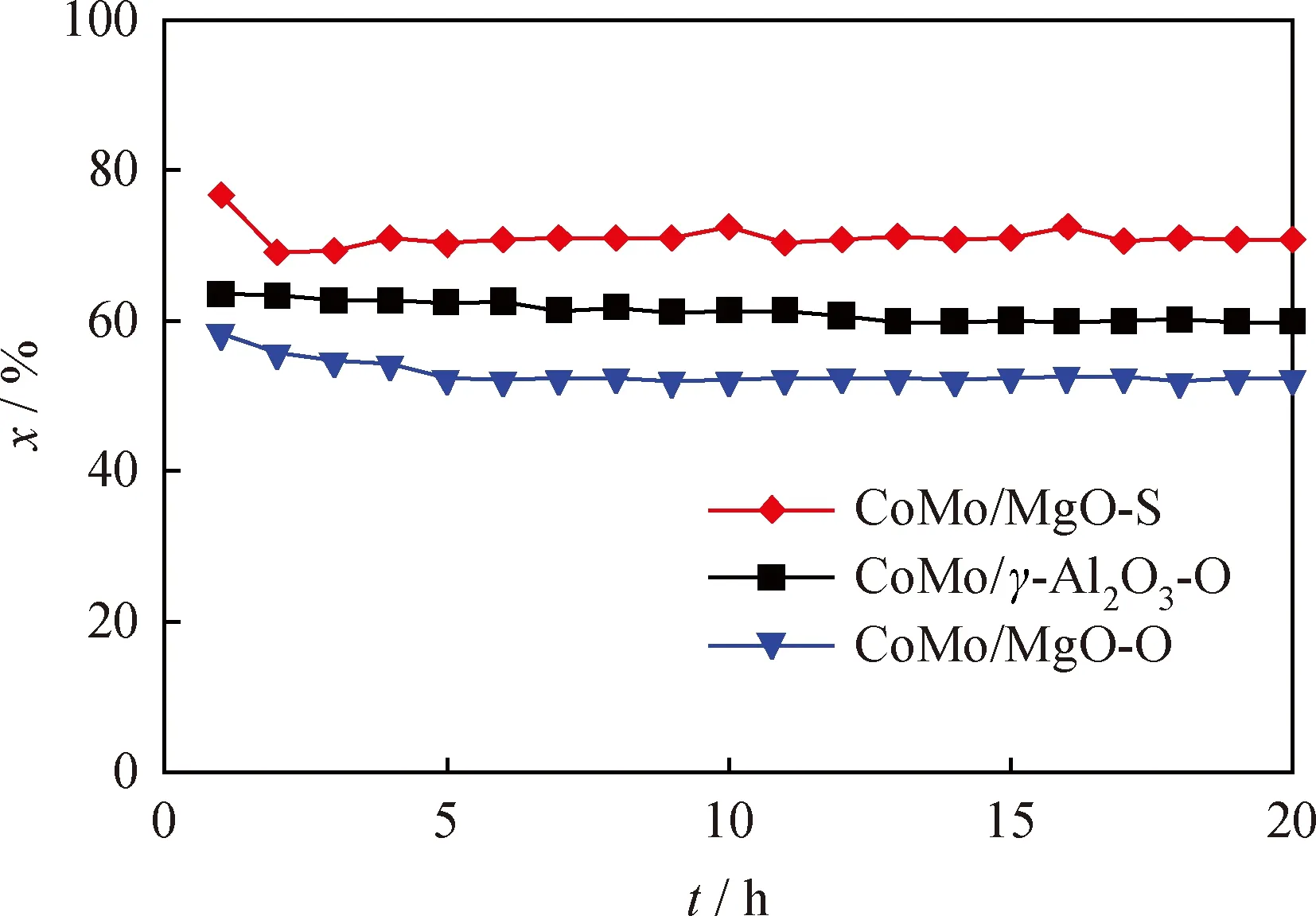
圖6 CoMo/MgO-S、CoMo/γ-Al2O3-O和CoMo/MgO-O催化劑上4,6-DMDBT的加氫脫硫活性反應中轉化率(x)隨反應時間(t)的變化曲線Fig.6 The conversion (x)vs t in the HDS reaction of 4,6-DMDBT over CoMo/MgO-S,CoMo/γ-Al2O3-O and CoMo/MgO-O catalystsp(H2)=5 MPa;T=300 ℃;m(Catalyst)=0.3 g;w0(4,6-DMDBT)=0.47%;qν(H2)=50 mL/min;MHSV=45 h-1
圖7為催化劑的本征活性比較,是在消除了內外擴散的前提下進行的比較(分別利用Weisz-Prater(CWP)和Mears(CM)判據消除內、外擴散傳質對催化反應的影響)[28]。計算得到CoMo/MgO-S催化劑的反應速率常數(kHDS)和催化轉化頻率(TOF)分別為0.094×10-6mol/(s·g)和2.2 h-1,而CoMo/MgO-O催化劑的kHDS和TOF分別為0.062×10-6mol/(s·g)和1.7 h-1。顯然,CoMo/MgO-S催化劑的本征加氫脫硫活性高于CoMo/MgO-O催化劑。
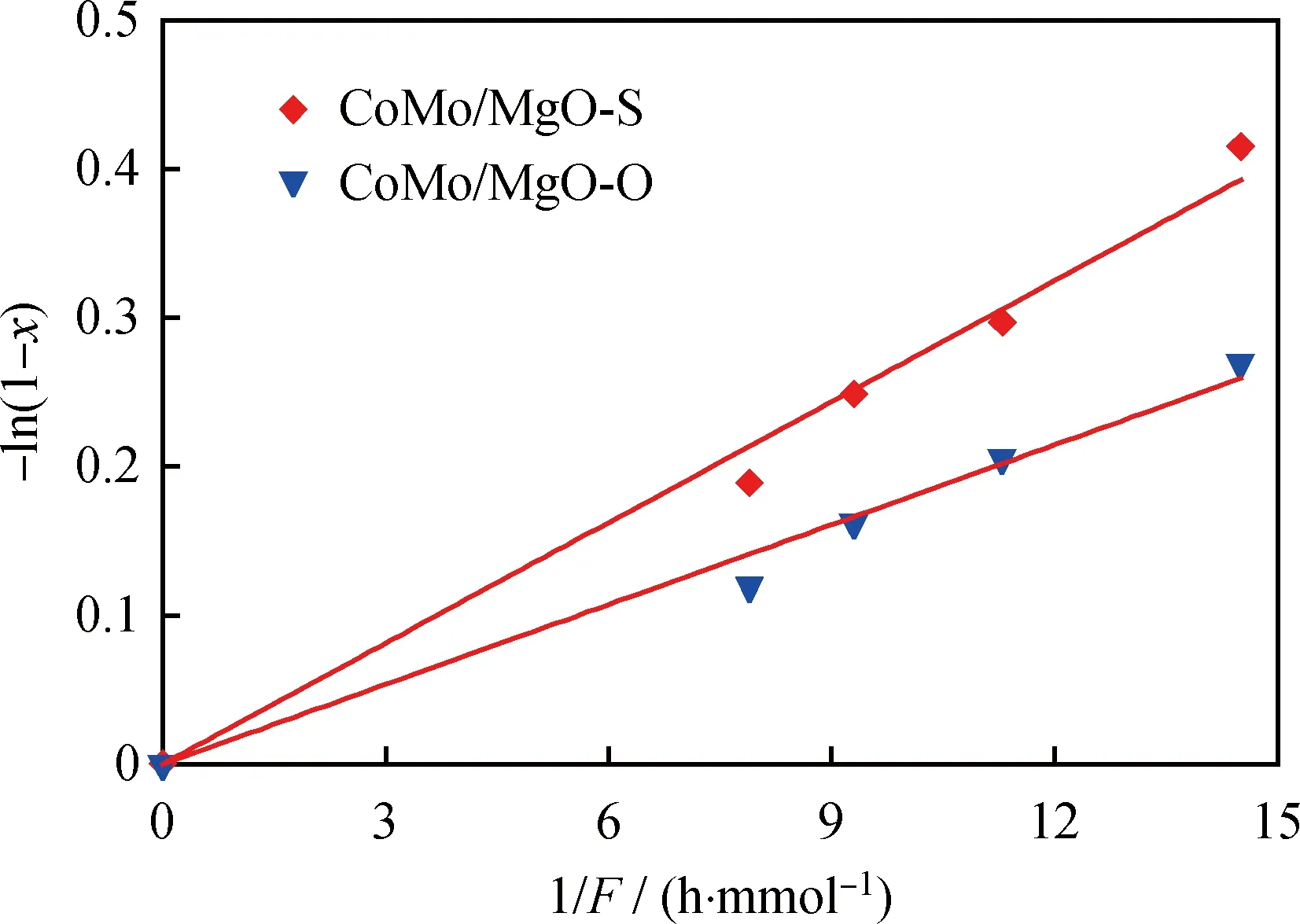
圖7 在CoMo/MgO-S和CoMo/MgO-O催化劑上-ln(1-x)與1/F的關系Fig.7 -ln(1-x)vs 1/F over CoMo/MgO-S and CoMo/MgO-O catalystsp(H2)=5 MPa;T=300 ℃;m(Catalyst)=0.08 g;w0(4,6-DMDBT)=0.47%;qν(H2)=50 mL/min
催化劑的加氫脫硫性能與金屬硫化物活性相的形貌有密切關系,根據Daage等[27]提出的“Rim-Edge”模型,Rim活性位分布在多層MoS2活性相的頂層和底層,是加氫和氫解的活性中心;Edge活性位在兩層中間,是氫解活性中心。前期研究結果也表明提高催化劑的加氫性能有利于4,6-DMDBT的 脫除[6]。通過TEM表征分析可知,與CoMo/MgO-O 催化劑相比,在CoMo/MgO-S催化劑上具有更多的多層MoS2相,形成了更多的加氫位點。XPS分析結果表明,與CoMo/MgO-O相比,在CoMo/MgO-S催化劑表面上有更多的Mo物種轉化成MoS2活性相,同時有更多的Co參與形成CoMoS活性相。盡管CoMo/MgO-O催化劑的孔徑大于CoMo/MgO-S催化劑,在反應過程中有利于4,6-DMDBT大分子的擴散,但是CoMo/MgO-S催化劑表面暴露的活性相要多于CoMo/MgO-O催化劑,使得4,6-DMDBT更容易與活性相發生吸附、催化反應,對催化性能的提高起到重要作用。此外,CoMo/MgO-S催化劑的表面積要高于CoMo/MgO-O催化劑,促進了反應分子與MoS2活性相接觸。因此,CoMo/MgO-S催化劑的加氫脫硫性能優于CoMo/MgO-O催化劑。
此外,與傳統CoMo/γ-Al2O3-O相比,催化劑的活性由大到小的順序為CoMo/MgO-S、CoMo/γ-Al2O3-O、CoMo/MgO-O。根據前期研究結果,MgO載體表面堿中心的強度和數量均優于γ-Al2O3載體[17]。當以鉬酸銨為前軀體制備催化劑時,酸性的Mo氧化物與堿性的MgO載體產生較強的相互作用,形成作用力較強的Mo—O—Mg鍵。與CoMo/γ-Al2O3-O相比,在CoMo/MgO-O上更容易形成加氫活性較低的單層MoS2活性相,不利于 4,6-DMDBT 的脫除。當以含硫的(NH4)2MoS4為前軀體時,MoS42-不存在與MgO載體表面形成Mo—O—Mg的條件,而容易硫化形成加氫活性較高的多層MoS2活性相,而在 CoMo/γ-Al2O3-O 催化劑上主要是加氫活性較低的單層的MoS2活性相[28]。因此,CoMo/MgO-S催化劑具有相對較高的加氫脫硫活性。
3 結 論
以四硫代鉬酸銨為前軀體制備了CoMo/MgO-S催化劑,在含硫大分子4,6-DMDBT的加氫脫硫反應中呈現了較好的催化性能,其活性要優于鉬酸銨為前軀體制備的CoMo/γ-Al2O3-O和CoMo/MgO-O催化劑。這主要是因為以四硫代鉬酸銨為前軀體制備的催化劑上,硫化過程中有利于Mo物種還原硫化形成更多的加氫性能較高的多層MoS2活性相。此外,CoMo/MgO-S催化劑具有較大的比表面積,增加了其與反應物的接觸面積。
猜你喜歡
課堂內外·初中版(科學少年)(2025年1期)2025-02-28 00:00:00
課堂內外·初中版(科學少年)(2025年2期)2025-02-28 00:00:00
英語世界(2023年10期)2023-11-17 09:18:18
科學大眾(中學)(2019年3期)2019-05-17 10:04:30
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
汽車觀察(2018年10期)2018-11-06 07:05:26
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
