大豆分離蛋白酶促聚集行為的表征
2019-09-06 07:52:46張志國
食品科學 2019年16期
關鍵詞:結構
曾 祺,張志國*
(齊魯工業大學(山東省科學院)食品科學與工程學院,山東 濟南 250353)
大豆分離蛋白(soybean protein isolate,SPI)由于其優異的營養學特性與加工特性,被廣泛運用于現代食品工業中[1]。SPI主要由β-伴大豆球蛋白(7S)和大豆球蛋白組成(11S)[2],這些蛋白通過氫鍵與分子內二硫鍵形成了穩定的球型結構[3],所以相較于其他蛋白而言,SPI穩定的球形結構阻礙了SPI分子間的相互作用,導致天然狀態下其聚集性較差,為滿足食品加工中各類食品的需求,需要改善SPI的聚集特性,酶法改性得益于其溫和安全的反應條件與安全高效的操作過程,已成為蛋白質食品資源的重要改性方式[4-6]。Zang Xiaodan等[7]報道了木瓜蛋白酶與植酸酶混合處理可提升SPI的乳化性能,Kuipers等[8]發現酶處理可拓寬SPI的冷凝膠條件。酶解處理不僅提高了SPI的功能性質,并且增強了食品的營養特性,因此,SPI的酶法改性受到了食品工業的廣泛關注。適當條件下的酶處理可引起SPI分子構象的改變,從而引起蛋白分子間作用力的一系列變化,導致SPI聚集特性改變。藍色刺孢霉是一種從紅曲米中分離得到的一種具有產天冬氨酸蛋白酶(aspartic protease,APs)能力的霉菌[9],APs可對SPI進行水解[10],改善其功能特性。本研究通過研究酶解后SPI組分、結構的變化,結合激光掃描共聚焦顯微鏡(laser scanning confocal microscopy,LSCM)與原子力顯微鏡(atomic force microscope,AFM)表征SPI的酶誘導聚集過程與聚集體的性質,揭示酶解后組分變化與結構特性的變化與SPI聚集特性之間的相關性,為SPI聚集特性在食品中的運用提供理論依據及參考。
1 材料與方法
1.1 材料與試劑
脫脂大豆粉由山東省禹王有限公司提供,Quambalaria cyanescens APs為實驗室自制(317.46 U/mL),其余試劑均為分析純。
1.2 儀器與設備
pHS-3E型pH計 上海佑科儀器儀表有限公司;UV759紫外-可見光分光光度計 上海佑科儀器儀表有限公司;電泳儀 美國Bio-Rad公司;F-4500型熒光分光光度計 日立高新技術公司;J-815型圓二色光譜儀日本Jasco分析儀器公司;Leica SP8型LSCM 德國萊卡生物系統;Multimode8 型AFM 德國布魯克公司。
1.3 方法
1.3.1 SPI的制備
SPI是由山東禹王有限公司提供的脫脂大豆粉為原料制得。制備步驟如下:將脫脂大豆粉懸浮于10 倍體積的蒸餾水中,用2 mol/L NaOH溶液調節pH值至8.0,室溫攪拌2 h后,置于離心機中以13 000×g離心20 min,收集上清液;上清液用2 mol/L HCl溶液調節pH值至4.5,隨后13 000×g離心15 min,收集沉淀,即為SPI。樣品凍干后,在4 ℃貯存備用。
1.3.2 SPI水解度的測定
采用pH-stat法,根據Adler-Nissen[11]的方法測定SPI在不同水解時間下的水解度,稱取一定量的SPI,用pH 7.0、10 mmol/L的磷酸鹽緩沖液配制為最終質量濃度5 mg/mL的SPI分散液,均質后的體系中加入Quambalaria cyanescens APs(質量分數1%),當所需的酶解時間達到后,加入2 mL的1 mg/mL Pepstatin A溶液終止酶解反應,滴定起始pH 8.0,水解過程中不斷加入0.5 mol/L的NaOH溶液以維持體系pH值穩定在8.0,水解度表示為在水解過程中破壞肽鍵與天然蛋白質中肽鍵總量之間的比值,由下式計算:
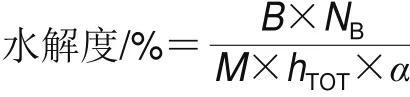
式中:B為反應過程中消耗堿量/mL;NB為堿濃度/(mol/mL);α為α-氨基酸在體系中的平均解離度,本實驗條件下取0.93[11];M為被酶解蛋白的總質量/g;hTOT為每克蛋白質底物具有的肽鍵物質的量,本實驗條件下為7.8 mmol/g[11]。
1.3.3 濁度的測定
采用UV759紫外-可見光分光光度計測定SPI在不同水解時間660 nm波長處光密度,以此表示濁度變化,表征SPI水解過程中聚集程度。
1.3.4 十二烷基硫酸鈉-聚丙烯酰氨凝膠電泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis,SDSPAGE)分析
采用Leammli[12]建立的不連續的電泳系統,將12%分離膠溶液灌注到膠室中,加膠高度為整個膠室的3/4。待分離膠完全聚合,除去分離膠膠面的水封層后,將配好的3%濃縮膠溶液灌注到分離膠的上方,至距凹玻璃板上方0.5 cm處,快速插入進樣梳,待濃縮膠完全聚合后,將主體放入電泳槽內。并倒入適量的pH 8.3的電極緩沖液,準確稱取0.001 g冷凍干燥后的不同酶解時間SPI置于EP管中,加入1 mL包含0.05 mol/L pH 8.0的Tris-HCl溶液、1% SDS、10%甘油、0.02%溴酚藍和1% β-巰基乙醇的還原性樣品溶解液,在沸水浴中加熱3 min,冷卻至室溫后加樣。加入10 μL樣品溶液和Marker,加至凝膠凹型樣品槽的底部。初始電流為15 mA,待樣品進入分離膠后,將電流調至30 mA,當溴酚藍染料距凝膠底部1 cm時停止電泳,將膠片取出固定、染色、脫色。
1.3.5 內源熒光光譜分析
將不同酶解時間處理后的SPI用10 mmol/mL pH 7.0磷酸鹽稀釋至最終質量濃度為0.05 g/mL,為減少SPI內部酪氨酸對熒光光譜的貢獻,設置激發波長為280 nm,在300~500 nm波長范圍內掃描檢測SPI內源性熒光強度的變化,掃描速率240 nm/min,激發和發射狹縫寬均為5 nm。
1.3.6 圓二色光譜分析
將不同酶解時間處理后的SPI用超純水稀釋至0.05 g/mL,以超純水為背景去除背景噪音,在溫度25 ℃、掃描速度100 nm/min、帶寬1.0 nm、反應時間0.50 s條件下測定SPI在遠紫外區(185~245 nm)的圓二色性。利用Applied Photophysics軟件表征SPI的二級結構包括α-螺旋、β-折疊、β-轉角和無規卷曲含量的變化。
1.3.7 LSCM觀察
將酶解后的SPI酶解液稀釋至1 mg/mL,取1 mL稀釋后的SPI加入10 μL 0.2 g/100 mL Rhodamine B染料,取20 μL染色后的SPI溶液滴加于凹槽載玻片中,蓋上蓋玻片,并用指甲油密封以防止加熱過程中溶液揮發,在40 ℃保溫2 h后,置于4 ℃保藏12 h。用Leica SP8型LSCM觀察SPI的聚集狀態。激發光568 nm,物鏡參數40×/NA 0.85。
1.3.8 AFM觀察
采用輕敲模式對不同酶解時間處理后的SPI微觀聚集形態進行觀察,探針微懸臂長度為180 μm,針尖曲率半徑為10 nm,針尖為商用氮化硅針尖,力常數為2.8 N/m。所有圖像只經過自動平滑處理,以消除慢掃描方向上的低頻噪音。每個樣品隨機選取6 個不同的區域,并用圖像分析軟件對SPI聚集體的微觀形態進行表征。
1.4 統計及分析
每組數據至少重復3 次,利用Origin 8.5軟件處理數據作圖,利用SPSS V17.0軟件對數據進行ANOVA差異顯著性分析及t檢驗。
2 結果與分析
2.1 不同酶解時間SPI的水解度與濁度
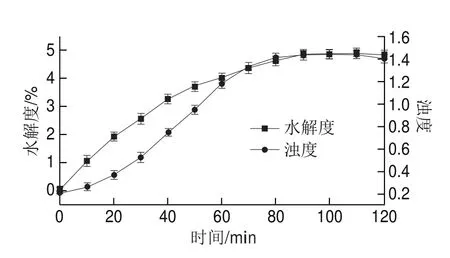
圖1 SPI的水解度與濁度變化Fig. 1 Change in DH value and turbidity of SPI
由圖1可知,SPI的水解度與濁度隨著酶解時間的延長而增加,通過SPI水解度的變化曲線可知,SPI的水解速度隨酶解時間的延長而減緩,并且在90 min左右到達酶解過程終點;由SPI的濁度變化曲線可知,SPI的聚集程度隨酶解時間延長而增大,但相較于SPI的水解程度,SPI的聚集行為存在一定的滯后,黃蘇[13]研究木瓜蛋白酶誘導的SPI酶促聚集行為,也發現相似的SPI聚集行為較于水解進程的滯后,可能由于水解初期所引起的SPI聚集驅動力的變化不足以克服SPI分子間保持的斥力,從而導致酶解SPI聚集行為的滯后現象。
2.2 不同酶解時間SPI的SDS-PAGE分析

圖2 不同時間酶處理后SPI的SDS-PAGE分析Fig. 2 SDS-PAGE analysis of SPI with different AP treatment times
SPI主要由7S和11S球蛋白組成,7S球蛋白主要由α(58~83 kDa)、α’(58~77 kDa)、β(42~53 kDa)3 個亞基結合而成[14-16],11S球蛋白由6 個亞基組成,每個亞基包含1 條酸性肽鏈和1 條堿性肽鏈,這些亞基通過二硫鍵連接起來,酸性肽鏈和堿性肽鏈的分子質量為31~45 kDa和18~20 kDa[17-18]。由圖2可知,天然SPI在40~55 kDa范圍內具有清晰蛋白條帶,此部分為SPI 7S組分中的β亞基(42~53 kDa),隨著酶處理時間延長,β亞基條帶逐漸消失,同時在37 kDa左右出現了新蛋白條帶的多肽,表明7S球蛋白中的β亞基被解離,并形成了分子質量大約為37 kDa的組分;相似的,在32 kDa的11S中酸性多肽鏈的條帶逐漸變淡直至消失,說明酶處理使得SPI中7S、11S中的亞基解離以及重排;相比之下,7S組分亞基的解離與11S相比較為明顯,可歸結于11S亞基的連接主要是通過二硫鍵結合,二硫鍵的鍵合方式在一定程度上阻礙了APs對11S組分的水解[13]。
2.3 內源熒光光譜表征SPI的結構變化
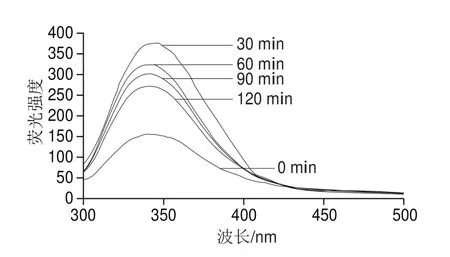
圖3 SPI的內源熒光光譜變化Fig. 3 Change in intrinsic fl uorescence emission spectrum of SPI with different AP treatment times
SPI的內源熒光主要由分子內的色氨酸殘基貢獻,色氨酸殘基所處環境的變化將會引起其熒光光譜的變化,因此,基于色氨酸殘基的內源熒光光譜常被用于蛋白相對構象變化的表征[19]。由圖2可知,APs酶處理組的SPI的熒光強度均高于對照(0 min),這是由于在天然狀態下的SPI具有穩固的球形結構,由于疏水水合作用,色氨酸等疏水性基團被包裹在蛋白結構的內部,隨著水解程度的增加,SPI相對構象發生變化,將色氨酸殘基等疏水性基團暴露于外部的親水環境中,從而導致了SPI熒光強度的變化。不僅如此,在酶處理過程中,SPI熒光強度隨酶解時間(30~60 min)上升而下降并且伴隨著峰位的輕微藍移(344~340 nm),這種漸進的熒光淬滅現象可以歸結于蛋白結構的部分恢復(或)及其聚集行為使暴露的色氨酸殘基恢復到疏水環境中[20];此外,臨近色氨酸蛋白分子間二硫鍵的形成也可以通過電子轉移過程導致類似熒光淬滅現象[21]。因此,APs酶解處理可使SPI相對構象改變及其內部疏水基團暴露,蛋白疏水基團的暴露可導致更強烈、更廣泛的疏水相互作用,從而誘導了SPI的聚集[22]。
2.4 圓二色光譜表征SPI二級結構變化
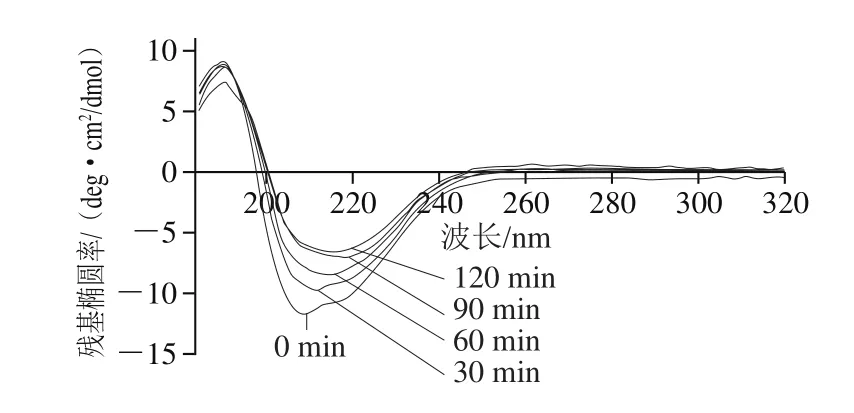
圖4 SPI遠紫外圓二色光譜的變化Fig. 4 Changes in far-UV CD spectrum of SPI with different AP treatment times
圖4 顯示,對照組(0 min)SPI的圓二色光譜在209 nm處顯示出一個清晰的負峰,在190 nm處顯示出一個正峰,這表明SPI具有高度有序的典型α+β結構[23],并且β-折疊結構含量占優勢[24],APs酶解處理導致SPI二級結構顯著性變化,隨著酶解時間的延長,SPI的負峰向長波長方向逐漸偏移并且伴隨著明顯的熒光強度升高;SPI的正峰未發現明顯偏移但其強度也有輕微下降。通過Applied Photophysics計算機軟件分析光譜得到SPI酶處理過程中二級結構含量的變化,酶處理0、30、60、90、120 min的SPI的α-螺旋含量分別為:(7.0±0.12)%、(10.3±0.05)%、(12.5±0.13)%、(15.7±0.07)%、(16.4±0.09)%;β-折疊含量分別為:(51.9±0.08)%、(42.8±0.09)%、(4 0.3±0.0 7)%、(3 9.2±0.0 4)%、(38.4±0.16)%;無規卷曲結構含量變化不顯著(P>0.05)。實驗結果表明APs酶處理導致了SPI二級結構中α-螺旋含量的上升及β-折疊含量的下降,類似的結構變化現象也出現在SO2誘導的葡萄酒中類奇異果甜蛋白的聚集行為中[25],根據Kim等[26]的研究,具有相同氨基酸序列的α-螺旋結構相較于β-折疊結構具有更高的聚集傾向。因此,蛋白二級結構中α-螺旋含量的增加有助于提升蛋白的聚集特性,從而引發蛋白的聚集行為。
2.5 LSCM分析
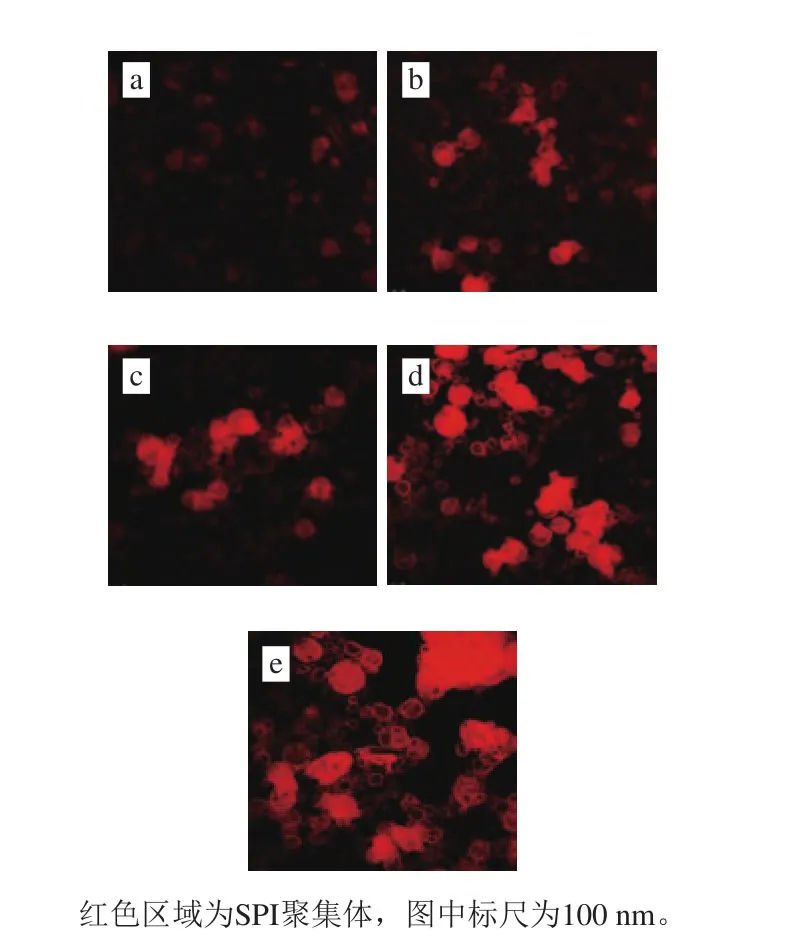
圖5 SPI酶誘導聚集行為的LSCM圖像Fig. 5 CLSM images of SPI aggregation behavior
如圖5所示,天然狀態下的SPI呈圓球狀,通過靜電相互作用與疏水作用穩定分散在溶液中,與Rhodamine B結合后熒光較弱。Rhodamine及其衍生物是屬于咕噸類的堿性染料,第3芳環(簡稱底環)可通過活性基與蛋白質、核酸等生物大分子及組成單體分子中的—NH2、—OH、—SH等形成共價鍵[27-29],使其熒光特性發生變化;在天然SPI結構中,—NH2與—SH基團大部分被包埋在SPI結構的內部,從而導致圖A中熒光強度較低;隨著酶處理時間的延長,SPI結構的變化導致更多基團與Rhodamine B染料結合,使其熒光強度上升(圖5B~E)。酶處理誘導了SPI的聚集行為,從圖5可以直觀地觀察到,隨著酶處理時間的延長,SPI形成了大小不均并且不連續的的聚集體,說明APs誘導的SPI聚集行為未能使SPI分子間產生足夠的共價交聯形成連續的凝膠體系,其聚集的主要驅動力還是由于SPI結構變化所導致的疏水相互作用以及靜電作用力等非共價作用力[29-31]。
2.6 AFM分析
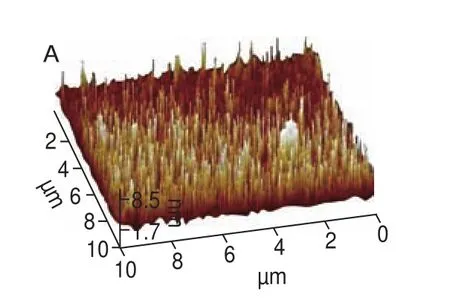
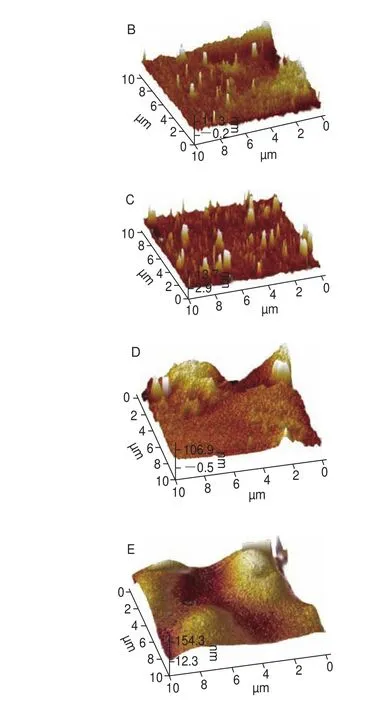
圖6 SPI在不同酶處理時間下聚集體的AFM圖像Fig. 6 AFM images of SPI aggregates with different enzyme treatment times
圖6 顯示了酶處理過程中由AFM觀察到的SPI微觀表面形態,未經酶處理的SPI主要以單體形式存在,簡單地吸附在云母片表面。當酶處理時間在0~60 min時,SPI分子逐漸開始形成較小、較為分散的聚集體(圖6B、C),此結果與LSCM觀察的結果相一致,當酶解時間進一步從60 min延長至120 min時,SPI間的聚集程度明顯增加,逐漸產生橫向融合,形成如圖6D、E的團狀不連續的聚集體,并在云母片表面形成了不規則的堆積結構。利用Nanoscope analysis軟件對SPI酶誘導聚集過程中的三維結構進行分析,不同酶處理時間下的SPI的平均表面粗糙度(Ra)分別為1.01、6.84、11.6、40.9、162 nm,Ra作為統計學的基本參數,能夠反映樣品表面粗糙程度的大小,即聚集程度的大小,Ra的增加說明SPI隨著酶處理時間的延長,其宏觀聚集程度增大,從而在云母片表面形成了不連續、不規則的SPI聚集體。
3 結 論
研究表明,APs處理可誘導SPI的聚集行為。SPI的水解度與濁度在酶處理過程中隨處理時間的延長而增大,表明SPI的解離程度與聚集程度隨酶處理時間的延長而增大;酶處理導致了SPI亞基的解離,將大分子質量SPI水解為具有較小分子質量的組分,其中7S組分較11S的解離程度較大。酶處理還導致SPI天然構象的改變,使埋藏在分子內部的疏水性基團及疏水性氨基酸暴露于更加親水的外部環境中,造成了SPI分子間更強、更廣泛的疏水相互作用;酶處理后的SPI結構中具有更高聚集傾向的α-螺旋含量增加,同時使SPI中對其二級結構穩定具有重要作用的β-折疊含量下降,增加了SPI分子的聚集傾向。APs誘導的SPI聚集行為并沒有形成高度有序的、連續的三維凝膠結構,聚集體很大程度依賴于非共價作用力形成不規則的堆積結構,其微觀結構表現為不連續、不規則的團狀聚集體。
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
