TLC-UPLC-MS/MS 法檢測網(wǎng)紅保健品中非法添加的8 種化學(xué)藥物
2019-09-06 07:53:22曹曉琴方振峰周國勇陳中強(qiáng)
食品科學(xué) 2019年16期
曹曉琴,方振峰,張 濤,周國勇,陳中強(qiáng),施 璐*
(江漢大學(xué)醫(yī)學(xué)院,湖北 武漢 430056)
目前,一些不法廠商為謀取經(jīng)濟(jì)利益,盲目追求療效,在網(wǎng)絡(luò)銷售的保健食品中非法添加化學(xué)藥物,變“禁藥”成“網(wǎng)紅藥”,嚴(yán)重危害人民群眾的生命健康安全[1]。其中,酚酞和西布曲明是網(wǎng)紅減肥藥中常見的添加物[2-3],一些商家還將麻黃堿和咖啡因聯(lián)合作為減肥的輔助藥物添加[4-5];西地那非是網(wǎng)紅補(bǔ)腎壯陽類保健品中常見的添加物[6];洛伐他汀和辛伐他汀是網(wǎng)紅降脂類保健品中常見的添加物[7];苯乙雙胍是網(wǎng)紅降糖類保健品中常見的添加物[8]。同時(shí),日常檢驗(yàn)過程中,還發(fā)現(xiàn)一些減肥類、降血脂類和潤腸通便類保健食品在功能宣稱和非法添加的藥物類型上往往具有交叉,既能迅速達(dá)到所宣稱的功效,又規(guī)避了非法添加化學(xué)藥物的檢測[9-10],而網(wǎng)售藥品和保健品作為一種新生事物,監(jiān)管體系還不健全,因此急需針對網(wǎng)紅保健品建立全面有效的檢測方法,打擊網(wǎng)上“多種類”、隱蔽性強(qiáng)的非法添加行為。
目前,保健品中非法添加西藥的常用檢測方法有薄層色譜(thin-layer chromatography,TLC)法[11]、高效液相色譜(high performance liquid chromatography,HPLC)法[12-14]、超高效液相色譜(ultra performance liquid chromatography,UPLC)法[15-16]、顯微傅里葉變換紅外光譜(micro fourier transform infrared spectroscopy,Micro FTIR)法[17]、表面增強(qiáng)拉曼光譜(surface-enhanced raman spectroscopy,SERS)法[18]、高效液相色譜-串聯(lián)質(zhì)譜聯(lián)用(high performance liquid chromatography-tandem mass spectrometry,HPLC-MS/MS)法[19-21]等。TLC和HPLC由于儀器性能限制問題,靈敏度低,容易造成假陽性的結(jié)果[11]。UPLC比傳統(tǒng)的液相色譜具有更高的分辨率和更快的分析速度,但是無法做到定量確證[15],HPLC-MS/MS法是目前監(jiān)測非法添加的有力工具,但是,文獻(xiàn)調(diào)研發(fā)現(xiàn),大部分監(jiān)測的對象僅集中在各自的小類里[22-23],而非法添加的網(wǎng)紅保健品為達(dá)到宣稱的功能療效,在非法添加的藥物類型上往往具有交叉,大大增加了檢測的難度。因此,本研究依據(jù)相關(guān)報(bào)道[24-26],選取了8 種具有減肥、降脂、通便功能的代表性藥物,同時(shí)也是從網(wǎng)紅商家處方便易得的一些代表性藥物,采用UPLC-MS/MS法進(jìn)行檢測。在前處理過程中,大部分文獻(xiàn)采取用甲醇或乙腈直接提取的方式進(jìn)行前處理[15,21],對一些復(fù)雜的基質(zhì)并沒有進(jìn)行針對性的處理,只是采用過濾膜去除雜質(zhì)干擾[24-26]。也有研究為避免基質(zhì)干擾,采用固相萃取法(solid-phase extraction,SPE)進(jìn)行前處理,但是操作過程復(fù)雜,成本較高[27-29]。
本研究將TLC法應(yīng)用到樣品前處理中,不僅避免了常規(guī)有機(jī)試劑提取方法容易造成干擾大、漏檢、假陰性的結(jié)果[9-11],也摒棄了文獻(xiàn)采用SPE法前處理成本高、較為復(fù)雜且不適合大量篩查的缺點(diǎn)[28-29],同時(shí)可以做到操作簡單,結(jié)果準(zhǔn)確。針對網(wǎng)紅保健食品中非法添加化學(xué)成分檢測方法的缺失和標(biāo)準(zhǔn)落后的情況,本實(shí)驗(yàn)將TLC法應(yīng)用到樣品前處理過程中,再聯(lián)用高靈敏度、高專一性的UPLC-MS/MS法,擬建立同時(shí)檢測網(wǎng)紅保健品中非法添加8 種不同類別的藥物(西布曲明、洛伐他汀、辛伐他汀、麻黃堿、咖啡因、酚酞、苯乙雙胍和西地那非)的TLC-UPLC-MS/MS方法。并采用該方法對市售45 種網(wǎng)紅保健食品進(jìn)行檢測,以期為打擊非法添加、藥品監(jiān)管及規(guī)范網(wǎng)絡(luò)市場提供技術(shù)支撐。
1 材料與方法
1.1 材料與試劑
鹽酸西布曲明(純度99.7%)、洛伐他汀(純度99.4%)、辛伐他汀(純度99.0%)、鹽酸麻黃堿(純度99.7%)、咖啡因(純度99.9%)、酚酞(純度99.9%)、鹽酸苯乙雙胍(純度99.7%) 中國食品藥品檢定研究院;枸櫞酸西地那非(純度99.5%) 美國Sigma-Aldrich公司;甲醇、乙腈、甲酸銨、甲酸、乙酸銨、乙酸(均為色譜純) 美國Fisher Chemicals公司;去離子水為Millipore公司超純水器制備;氯仿、氨水、氫氧化鈉、鹽酸(均為分析純)、羧甲基纖維素鈉(批號(hào):20161220) 國藥集團(tuán)化學(xué)試劑有限公司;GF254薄層硅膠板(200 mm×100 mm) 青島海浪硅膠干燥劑廠。
45 批保健食品樣品均來自網(wǎng)購。
1.2 儀器與設(shè)備
API 4500三重四極桿MS/MS 美國Applied Biosystems公司;UPLC 日本Shimadzu公司;Milli-Q超純水系統(tǒng) 美國Millipore公司;離心機(jī) 湖南湘立科學(xué)有限公司;Kinetex 100 RP-18色譜柱 美國Phenomenex公司。
1.3 方法
1.3.1 色譜條件
色譜柱(100 mm×2.1 mm,2.6 μm),柱溫:35 ℃;進(jìn)樣量:10 μL;流速:0.2 mL/min。流動(dòng)相梯度見表1。
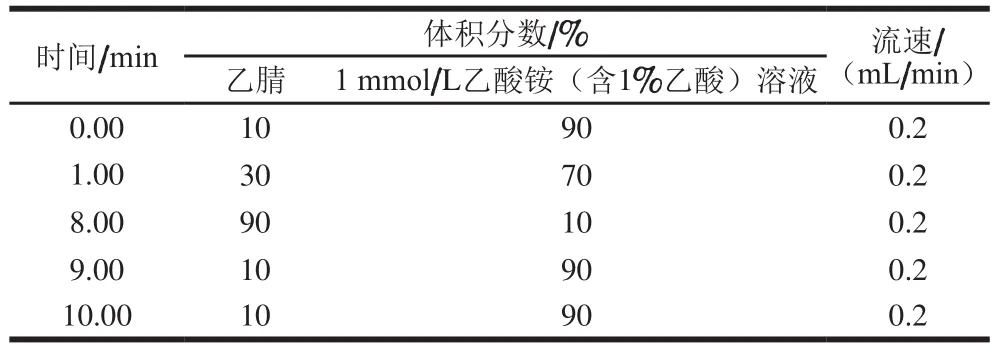
表1 UPLC梯度洗脫程序Table 1 Mobile phase gradient program of UPLC
1.3.2 質(zhì)譜條件
結(jié)合文獻(xiàn)[19-21]報(bào)道,本研究分別采用乙腈-水、5 mmol/L乙酸銨-乙腈、0.2 mmol/L乙酸-乙腈的溶劑體系(50∶50,V/V)配制10 μg/L的標(biāo)準(zhǔn)溶液,依次進(jìn)行質(zhì)譜條件的選擇。采用參數(shù)分別為:離子源:電噴霧電離(electrospray ionization,ESI)源;掃描方式:正離子模式進(jìn)行掃描;氣簾氣15 psi;霧化氣(Gas1):55 psi;輔助氣(Gas2):50 psi;離子噴霧電壓:5 500 V;離子源溫度:550 ℃;在多重反應(yīng)監(jiān)測(multiple reaction monitoring,MRM)狀態(tài)下,通過優(yōu)化離子對的碰撞能量和去簇電壓值,得到各目標(biāo)化合物的質(zhì)譜參數(shù)見表2。
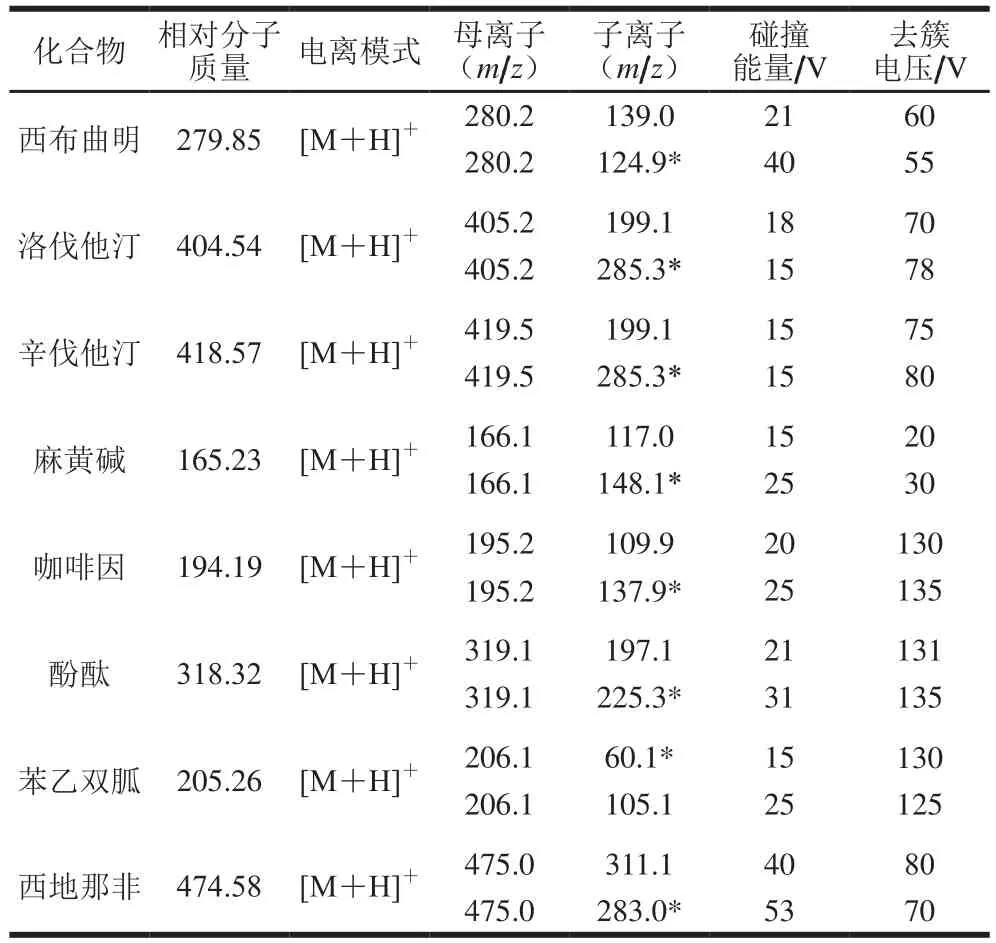
表2 8 種藥物在MRM模式下的母離子和子離子信息Table 2 Precursor/daughter ions information of the 8 drugs in the MRM mode
1.3.3 對照品溶液的制備
標(biāo)準(zhǔn)對照品貯備液的配制:精密稱取各對照品10.0 mg,用10 mL甲醇溶解、定容,分別制成質(zhì)量濃度為1.0 mg/mL標(biāo)準(zhǔn)貯備液,-20 ℃避光保存。
混合對照品標(biāo)準(zhǔn)工作液的配制:精密量取標(biāo)準(zhǔn)貯備液各0.1 mL,加入空白樣品提取液中,定容到10 mL,制成質(zhì)量濃度為10.0 μg/mL的混合標(biāo)準(zhǔn)工作液,4 ℃避光保存。
1.3.4 供試品的制備
片劑:隨機(jī)取同一批號(hào)的供試品20 片,研細(xì)為試樣;硬膠囊:隨機(jī)取同一批號(hào)的供試品20 粒,傾出所有內(nèi)容物,研細(xì)為試樣;軟膠囊:隨機(jī)取同一批號(hào)的供試品20 粒,將內(nèi)容物全部擠到同一離心管中,充分混勻后,作為試樣;口服溶液劑:隨機(jī)取同一批號(hào)的供試品10 支(瓶),取等量體積樣品到同一容器中混勻,作為試樣;其他固體類樣品(餅干、糖果、粉末、茶劑):隨機(jī)取同一批號(hào)的供試品10 份,研細(xì)或粉碎為試樣。
稱取上述處理好的試樣約1.0 g,置于5 mL離心管中,加入2 mL甲醇,超聲提取(功率143 W;頻率40 kHz)15 min后,4 000 r/min離心5 min,取上清液作為供試品溶液。
所有采集的樣品均采用建立的檢測方法進(jìn)行空白基質(zhì)的篩選,測定結(jié)果為無任何干擾峰的基質(zhì)作為空白基質(zhì),用于樣品添加實(shí)驗(yàn)。
1.3.5 前處理?xiàng)l件
本研究采用直接提取法、SPE法和TLC法分別進(jìn)行實(shí)驗(yàn)。
直接提取法采用文獻(xiàn)[21]方法,稱取1.3.4節(jié)處理好的試樣約1.0 g,置于5 mL 離心管中,加入2 mL甲醇,超聲提取(功率143 W;頻率40 kHz)15 min后,4 000 r/min離心5 min,取上清液,過0.22 μm濾膜上機(jī)檢測。
SPE法參考文獻(xiàn)[28-29]方法,采用Waters Oasis HLB SPE小柱1 mL/30 mg,依次加入甲醇3 mL和水3 mL活化后,經(jīng)1.3.4節(jié)制備好的供試品溶液上樣,再用甲醇-水(50∶50,V/V)3 mL淋洗,抽干后,用甲醇3 mL洗脫,收集洗脫液。氮?dú)獯蹈桑? mL流動(dòng)相復(fù)溶,過0.22 μm 濾膜,續(xù)濾液作為供試品溶液。
TLC法在文獻(xiàn)[17-18]基礎(chǔ)上進(jìn)行適當(dāng)調(diào)整,以硅膠GF254薄層板為固定相,以氯仿-甲醇-氨水(11∶1∶0.1,V/V)為展開劑,取制備好的供試品溶液點(diǎn)于薄層板上,點(diǎn)樣量為0.5 mL,展開后取出晾干,在紫外燈254 nm下檢視定位。將顯色的薄層挖下來放入1.5 mL EP管中,將薄層粉末連同EP管一同烘干后,用0.5 mL的甲醇溶解,超聲 5 min、高速離心3~5 min,取上清液,過濾膜上機(jī)檢測。
1.3.6 定量測定
空白樣品提取液中加入1.3.3節(jié)制備的對照品溶液制成混合標(biāo)準(zhǔn)溶液,在1.3.1節(jié)和1.3.2節(jié)的色譜和質(zhì)譜條件下進(jìn)樣分析,以各目標(biāo)物質(zhì)的響應(yīng)信號(hào)強(qiáng)度y為縱坐標(biāo),對應(yīng)藥物的濃度為橫坐標(biāo)繪制標(biāo)準(zhǔn)工作曲線。樣品溶液中各待測藥物響應(yīng)值均應(yīng)在測定的線性范圍內(nèi)。
1.4 統(tǒng)計(jì)學(xué)分析
在優(yōu)化條件和實(shí)際樣品測定時(shí),平行實(shí)驗(yàn)重復(fù)操作6 次,并采用SPSS 13.0(Chicago, IL, USA)統(tǒng)計(jì)軟件分析。基質(zhì)效應(yīng)通過t檢驗(yàn)考察[30]。
2 結(jié)果與分析
2.1 質(zhì)譜條件的選擇
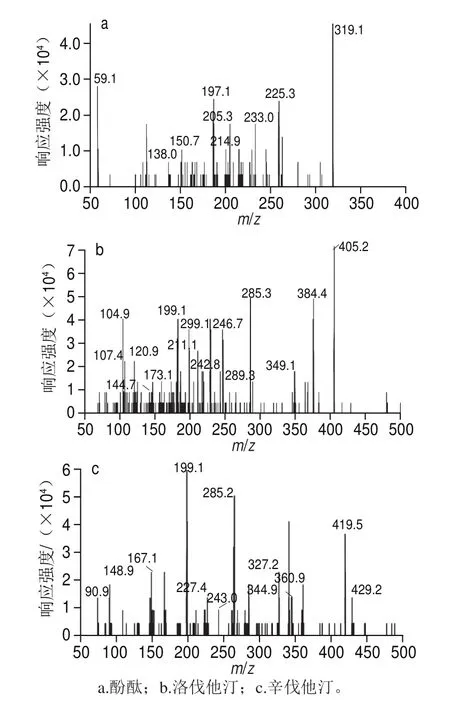
圖1 部分藥物的二級(jí)質(zhì)譜圖Fig. 1 Mass spectra of selected drugs
質(zhì)譜條件選擇時(shí),ESI的正負(fù)離子模式均被評(píng)估,結(jié)果發(fā)現(xiàn),待測物均可選擇[M+H]+離子作為母離子。這與許立等[9]報(bào)道一致。而洛伐他汀和辛伐他汀很容易加和成[M+Na]+模式,而且響應(yīng)很高,但其二級(jí)碎片離子穩(wěn)定性較差,而響應(yīng)較弱的[M+H]+模式在MRM下取得較好的響應(yīng),而且比較穩(wěn)定,因此選擇[M+H]+模式,這與宮旭等[21]報(bào)道的一致。根據(jù)馬微[3]和李濤[4]等報(bào)道,酚酞采用正負(fù)離子模式都可檢測,馬微等[3]認(rèn)為正離子響應(yīng)高于負(fù)離子模式,但是本研究發(fā)現(xiàn)負(fù)離子的質(zhì)譜響應(yīng)信號(hào)高于正離子模式下的響應(yīng)。在與其他藥物同時(shí)檢測時(shí),采用正負(fù)離子同時(shí)掃描的模式,響應(yīng)值明顯低于正離子掃描模式。所以綜合考慮,在流動(dòng)相中加入10 mmol/L乙酸銨溶液,使得正離子條件下的酚酞也獲得了較高的靈敏度,形成m/z 319的[M+H]+的準(zhǔn)分子離子峰。對于結(jié)構(gòu)相似的物質(zhì)如洛伐他汀和辛伐他汀,裂解成相同的子離子,但通過離子對的組合,可以實(shí)現(xiàn)良好的分離。總之,通過優(yōu)化各自的離子對,所有待測藥物都選擇了合適的母離子和子離子,部分藥物的二級(jí)質(zhì)譜圖見圖1。
2.2 色譜條件的選擇
2.2.1 色譜柱
根據(jù)文獻(xiàn)報(bào)道[20-22],結(jié)合選定的目標(biāo)化合物的性質(zhì),分別考察了Agilent Eclipse Plus C18柱(2.1 mm×100 mm,1.8 μm)、Waters BEH Shield RP-18(100 mm×2.1 mm,1.7 μm)色譜柱、Kinetex 100 RP-18柱(100 mm×2.1 mm,2.6 μm)等。結(jié)果表明Agilent Eclipse Plus C18柱分離時(shí)整體出峰較好,但麻黃堿、咖啡因色譜峰出現(xiàn)拖尾;Waters BEH Shield RP-18柱適合分離堿性強(qiáng)的物質(zhì),因此麻黃堿、咖啡因峰形更尖銳對稱,但是分離時(shí)酚酞和苯乙雙胍峰不能分離;Kinetex 100 RP-18柱對8 個(gè)物質(zhì)都能完全分離,各峰峰形也很尖銳對稱,分析時(shí)間較短。綜合考慮,最終選擇Kinetex 100 RP-18柱(100 mm×2.1 mm,2.6 μm)為本實(shí)驗(yàn)的色譜柱。
2.2.2 流動(dòng)相
對于反相色譜,通常采用甲醇-水、乙腈-水等二元溶劑體系作為流動(dòng)相進(jìn)行實(shí)驗(yàn),根據(jù)許立等[9]報(bào)道,正離子掃描的模式通常采用甲酸增強(qiáng)儀器響應(yīng),但是也有文獻(xiàn)采用加入不同比例的甲酸銨或乙酸銨取得很好的效果[21]。所以本研究分別比較了不同的溶劑配比對分離效果的影響,流動(dòng)相中依次加入甲酸、乙酸、甲酸銨、乙酸銨等離子化試劑進(jìn)行流動(dòng)相的選擇。結(jié)果發(fā)現(xiàn):以不同體積比的乙腈-水為流動(dòng)相時(shí)待測物的儀器響應(yīng)信號(hào)高于同比例的甲醇-水為流動(dòng)相。流動(dòng)相中加入乙酸銨可改善待測物的分離與峰形,正離子模式中加入甲酸可顯著提高待測物電離效果。這與許立等[9]的結(jié)果一致。所以最終選定乙腈和1 mmol/L乙酸銨(含1%乙酸)溶液作為流動(dòng)相。在選定的流動(dòng)相下各組分獲得良好的分離,通過優(yōu)化梯度洗脫程序,將不同極性的待測物的檢測時(shí)間縮短為10 min。8 種待測物的總離子流圖見圖2。
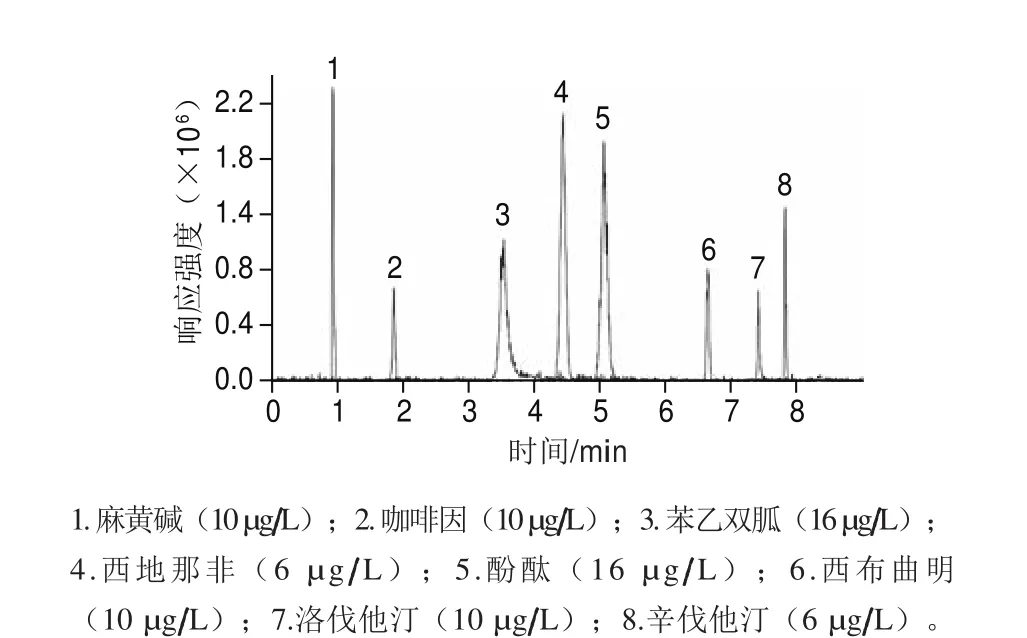
圖2 8 種待測物的總離子流圖(6~16 μg/L)Fig. 2 Total ion current chromatograms of the 8 compounds (6–16 μg/L)
2.2.3 柱溫的選擇
考察15、25、35、45 ℃ 4 種不同柱溫對分離度的影響。通過比較發(fā)現(xiàn)在4 種溫度條件下,分析總時(shí)間相差不大,但是在15 ℃時(shí),洛伐他汀和辛伐他汀分離度下降,兩峰會(huì)部分重疊。當(dāng)柱溫大于40 ℃時(shí),苯乙雙胍峰和酚酞峰因保留時(shí)間間隔縮短而未能分離,從而兩種物質(zhì)合為一個(gè)色譜強(qiáng)峰。柱溫35 ℃時(shí)的實(shí)驗(yàn)結(jié)果最佳(圖3)。因此本實(shí)驗(yàn)柱溫最終定為35 ℃。
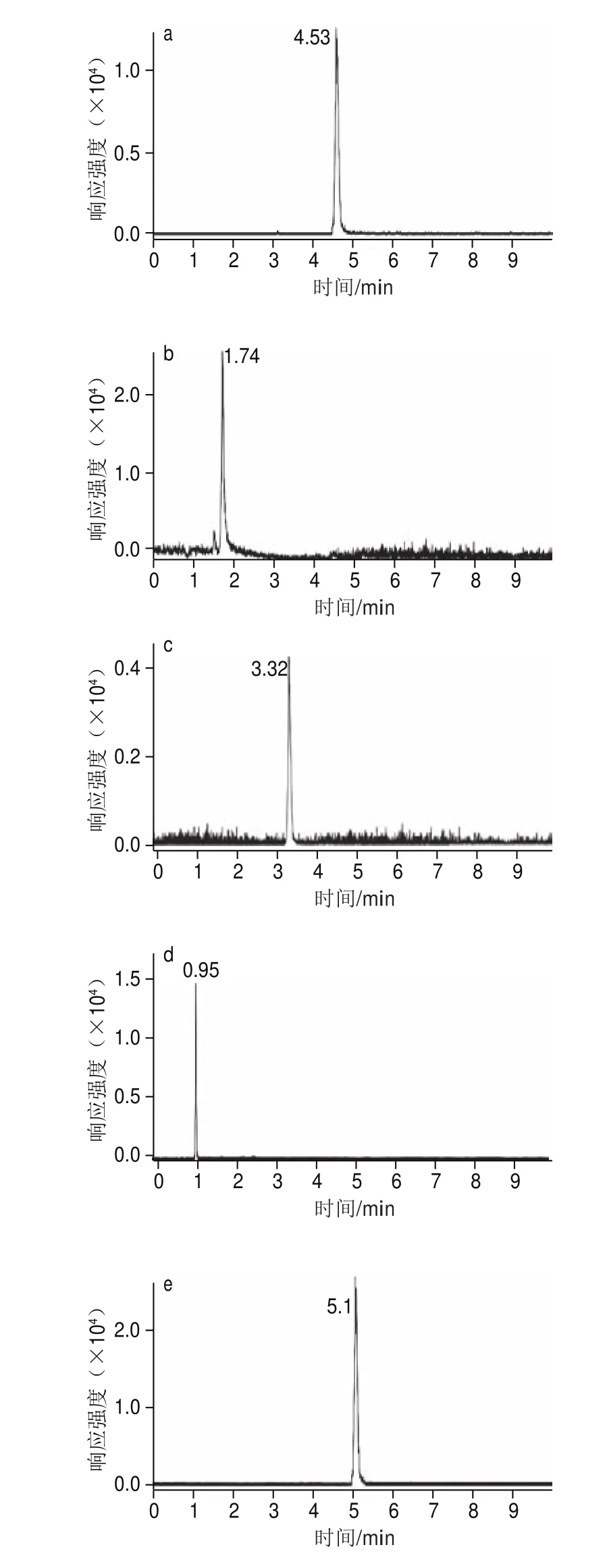
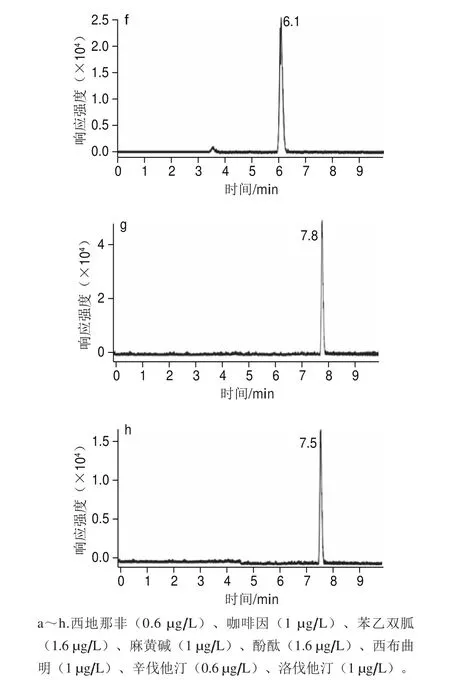
圖3 標(biāo)準(zhǔn)溶液中8 種化合物的定量色譜圖(0.6~1.6 μg/L)Fig. 3 Quantitative chromatograms of standard solutions of the 8 compounds (0.6–1.6 μg/L)
2.3 前處理方法的選擇
直接提取法和SPE法分別參考文獻(xiàn)[21]和文獻(xiàn)[17-18]進(jìn)行實(shí)驗(yàn)(如1.3.5節(jié)所述)。TLC法在文獻(xiàn)[17-18]的基礎(chǔ)上,對展開劑進(jìn)行了優(yōu)化,分別考察氯仿-甲醇-丙酮-冰醋酸(9∶2∶1∶0.5,V/V)和氯仿-甲醇-氨水(11∶1∶0.1,V/V)的展開效果,結(jié)果無明顯差異。考慮到配比的方便,最終確定采用的是氯仿-甲醇-氨水(11∶1∶0.1,V/V)作為展開劑(具體步驟見1.3.5節(jié))。
對3 種提取方法的提取效果進(jìn)行詳細(xì)比較,直接提取方法操作簡單快速,但是稀釋了藥物的濃度,會(huì)影響測定的靈敏度,而且對于一些復(fù)雜的樣品基質(zhì),只進(jìn)行簡單的提取,未經(jīng)凈化,上機(jī)樣液雜質(zhì)較多,容易導(dǎo)致色譜柱壓力波動(dòng),影響保留時(shí)間定性,影響數(shù)據(jù)的準(zhǔn)確性,而且極易污染離子源和檢測器[27]。SPE法在提取后進(jìn)行了凈化,采用SPE小柱進(jìn)一步對樣品進(jìn)行處理,消除了基質(zhì)干擾,但是SPE成本高且操作較為復(fù)雜。此外文獻(xiàn)認(rèn)為SPE會(huì)降低方法的回收率[27]。所以本研究將色譜分析中的TLC法應(yīng)用到樣品前處理中,既避免了傳統(tǒng)的液液萃取法容易造成干擾大、漏檢、假陰性的結(jié)果,也摒棄了SPE成本高且較為復(fù)雜的方式。通過對3 種提取方法提取回收率的比較(表3),結(jié)果顯示,采用TLC法前處理與SPE法取得同樣的提取效果,明顯優(yōu)于傳統(tǒng)的直接提取法,但TLC法比SPE法成本更低,操作更簡單,更易于實(shí)現(xiàn)大規(guī)模的檢測,最終本實(shí)驗(yàn)采用TLC進(jìn)行前處理取得較好的效果。
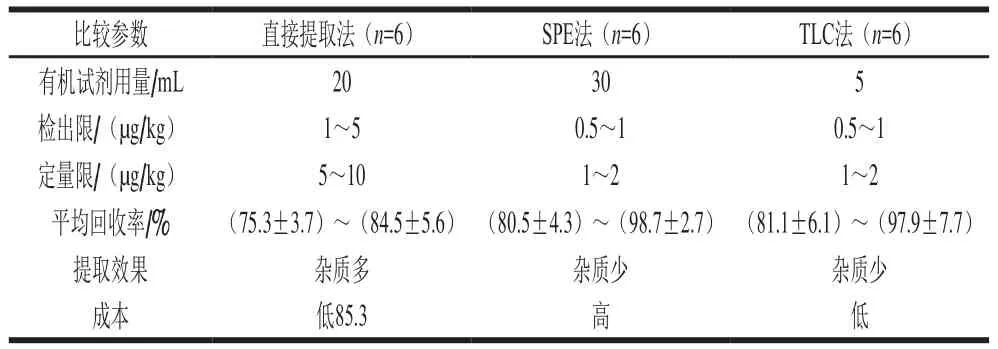
表3 3 種不同的前處理方法比較Table 3 Comparison of three extraction techniques
2.4 方法學(xué)考察結(jié)果
2.4.1 基質(zhì)效應(yīng)考察結(jié)果
采用t檢驗(yàn)考察基質(zhì)效應(yīng)[30]。經(jīng)實(shí)驗(yàn)得出8 種藥品在不同基質(zhì)中的t值均大于2.4。所以,本研究采用基質(zhì)匹配工作曲線進(jìn)行定量。
2.4.2 檢出限、定量限及線性范圍
本研究在文獻(xiàn)[20-22]的基礎(chǔ)上,利用配制成的空白樣品中加入混合標(biāo)準(zhǔn)溶液,按0.5、1.0、5.0、10、50、100 μg/L的水平添加,進(jìn)樣檢測,繪制標(biāo)準(zhǔn)工作曲線。以峰面積Y對進(jìn)樣質(zhì)量濃度X進(jìn)行線性回歸,以RSN為3計(jì)算檢出限,以RSN為10計(jì)算定量限)。由表4可知,8 種藥物在質(zhì)量濃度0.5~100 μg/L范圍內(nèi),線性關(guān)系良好,各組分的檢出限在0.09~0.29 μg/kg之間,定量限在0.31~0.79 μg/kg之間。
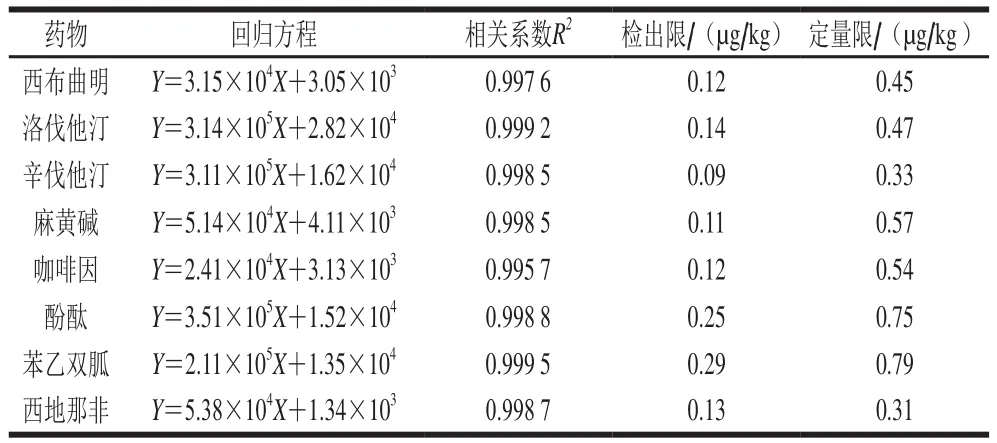
表4 8 種待測藥物的線性回歸方程、相關(guān)系數(shù)、檢出限及定量限Table 4 Regression equations, correlation coefficients, limits of detection and limits of quantization for the 8 drugs
2.4.3 準(zhǔn)確度和精密度結(jié)果
準(zhǔn)確度用回收率表示,分別在3 個(gè)質(zhì)量濃度水平上添加(1、2、4 倍定量限)。UPLC-MS/MS方法的專屬性非常強(qiáng),同類型樣品基質(zhì)的干擾可忽略,因此,本研究選取膠囊劑和口服液和其他固體制劑的代表粉末劑分別做回收率實(shí)驗(yàn)。如表5所示,對于膠囊劑和粉末劑,取其中陰性樣品(12號(hào)),稱取1 g樣品,精密加入對照品儲(chǔ)備溶液,最終使試樣中西布曲明、洛伐他汀、麻黃堿、咖啡因含量0.5、1.0、2.0 μg/kg;辛伐他汀和西地那非含量為0.3、0.6、1.2 μg/kg,酚酞和苯乙雙胍含量為0.8、1.6、3.2 μg/kg。對于口服液,取其中陰性樣品(8號(hào)),量取1 mL樣品,精密加入對照品儲(chǔ)備溶液,最終使試樣中西布曲明、洛伐他汀、麻黃堿、咖啡因含量為0.5、1.0、2.0 μg/kg;辛伐他汀和西地那非含量為0.3、0.6、1.2 μg/kg,酚酞和苯乙雙胍含量為0.8、1.6、3.2 μg/kg。對于軟膠囊劑,取其中陰性樣品(43號(hào)),稱取1 g樣品,精密加入對照品儲(chǔ)備溶液,最終使試樣中西布曲明、洛伐他汀、麻黃堿、咖啡因含量為1.0、2.0、4.0 μg/kg;辛伐他汀和西地那非含量為0.5、1.0、2.0 μg/kg,酚酞和苯乙雙胍含量為1.5、3.0、6.0 μg/kg。
按照前面優(yōu)化的前處理方法和分析測定方法操作,每個(gè)含量樣品重復(fù)5 次,通過連續(xù)5 d進(jìn)行同樣實(shí)驗(yàn),以回收率表示方法的準(zhǔn)確度,變異系數(shù)表示方法的精密度。不同樣品中添加8 種藥物的回收率及變異系數(shù)見表5,分析數(shù)據(jù)可知待測物在不同樣品中的平均回收率介于67.3%~101.1%,變異系數(shù)≤16.5%,這些數(shù)據(jù)均符合分析方法標(biāo)準(zhǔn)對準(zhǔn)確度和精密度的要求。

表5 不同樣品中添加8 種藥物回收率和變異系數(shù)Table 5 Recoveries and coefficients of variation for the 8 drugs from different spiked samples
2.5 樣品測定結(jié)果
取網(wǎng)購保健食品樣品,按1.3節(jié)制備方法制備,分別進(jìn)樣測定,根據(jù)定量監(jiān)測離子對的離子流圖中色譜峰面積,采用外標(biāo)法計(jì)算樣品中各化學(xué)成分的量。排除目標(biāo)藥物是由處方成分引入的,最后判定樣品中是否含有非法添加的藥物。
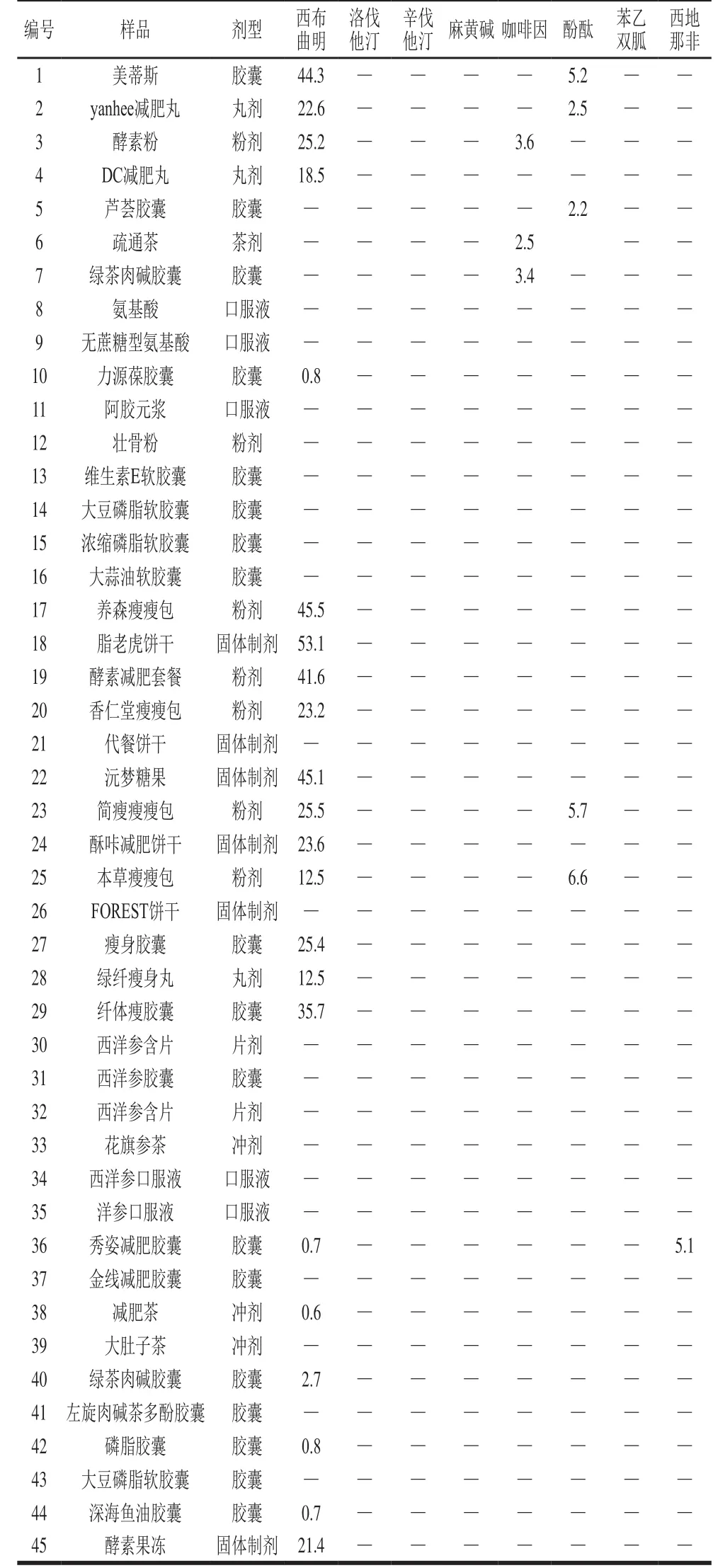
表6 45種網(wǎng)購樣品中8 種待測藥物的分析結(jié)果(n=6)Table 6 Results of detection of 8 drugs in 45 real samples (n= 6)
表6結(jié)果表明,45 批保健食品樣品中25 批樣品檢出含有目標(biāo)藥物。通過與樣品相應(yīng)的成分進(jìn)行比對,其中2 批樣品中所檢出的目標(biāo)藥物與處方固有成分一致,分別為疏通茶和綠茶肉堿膠囊檢出咖啡因。因?yàn)椴枞~中會(huì)有一定量的咖啡因,不屬于非法添加;另外23 批樣品檢出的目標(biāo)化合物均不屬于處方所標(biāo)示的成分,確定為非法添加陽性樣品,采用2.5節(jié)線性方程計(jì)算其中非法添加藥物的含量,結(jié)果表明:檢出率較高的是西布曲明(21 批,含量為0.6~53.1 mg/g),酚酞(5 批,含量為2.2~6.6 mg/g),咖啡因和西地那非分別有1 批。
3 結(jié) 論
本研究建立了TLC-UPLC-MS/MS法檢測網(wǎng)紅保健品中非法添加的8 種化學(xué)藥物的檢測方法。根據(jù)方法學(xué)考察結(jié)果,本方法專屬性強(qiáng),靈敏度高,可作為非法摻入化學(xué)成分的有效檢測方法。通過對網(wǎng)購的45 種網(wǎng)紅保健品進(jìn)行檢測,為打擊網(wǎng)絡(luò)非法添加,規(guī)范保健品市場,提供了技術(shù)支持。
猜你喜歡
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
海峽科技與產(chǎn)業(yè)(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
中國衛(wèi)生標(biāo)準(zhǔn)管理(2015年17期)2016-01-20 09:26:45
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
中國當(dāng)代醫(yī)藥(2015年17期)2015-03-01 02:03:39
中國衛(wèi)生標(biāo)準(zhǔn)管理(2015年13期)2015-01-26 21:05:38
