高效液相色譜-電感耦合等離子體質譜法測定配方食品中的VB12
2019-09-06 07:53:20朱曉玲彭青枝周陶鴻張小玲
食品科學 2019年16期
劉 杰,龔 蕾,朱曉玲*,彭青枝,張 莉,周陶鴻,江 豐,黃 茜,張小玲
(湖北省食品質量安全監督檢驗研究院,湖北省食品質量安全檢測工程技術研究中心,湖北 武漢 430070)
VB12又稱鈷胺素,是一種由含鈷的卟啉類化合物組成的B族維生素,對維持人體基本機能具有重要的作用[1-2]。VB12無法在人體內合成,且幾乎不存在于植物性食品中[3],因此,人體只能從動物性食物中獲得。由于人們的飲食結構變化,素食主義者增多[4],導致部分居民出現膳食營養不平衡問題,市面上由此大量涌現各類營養強化食品、保健食品等配方食品,在其中人為添加營養物質VB12,以滿足部分人群補充VB12的需求。但目前許多保健食品B族維生素片所添加的VB12含量并不規范,VB12過量攝入會對人體產生一定的毒副作用[5],必須規范控制這類產品中VB12的使用量,并對其中的VB12含量進行準確測定。
對于VB12的檢測,至今仍然是國際上食品分析的難題之一。原因主要有兩點:一是動物性食品中的VB12存在氰鈷胺、羥鈷胺、腺苷鈷胺和甲基鈷胺等多種結構形式[6],傳統的分析方法很難反映樣品中VB12的實際含量,通常需要采用氰化鉀或氰化鈉等劇毒試劑將鈷胺素異構體(羥鈷胺素、甲鈷胺素和5-脫氧腺苷鈷胺素等)轉化為較穩定的氰鈷胺素后再進行分析檢測,而目前尚未尋找到合適的劇毒試劑替代物,因此方法的適用性受限,難以推廣;二是VB12在食品中的含量極低,一般為10-9級,在強化食品中按照國家標準要求也僅約0.6~70 μg/kg[7],對分析方法的檢測靈敏度具有較高的要求。另外,VB12通常以VB12-內因子復合物的形式存在,充分提取和分離難度很大,樣品中其他化合物以及與VB12相近的水溶性維生素也會對其分離檢測造成干擾。
目前,食品中VB12檢測的國家標準主要有GB 5413.14—2010《嬰幼兒食品和乳品中維生素B12的測定》[8]和GB/T 5009.217—2008《保健食品中維生素B12的測定》[9],所采用的方法為微生物法和液相色譜法,微生物法檢測VB12的優勢在于具有較高的靈敏度,但缺點主要為其操作過程較繁瑣,對人員的技術要求較高,且檢測耗時,測定周期需要3 d;液相色譜法主要可用于VB12添加量較高的保健食品的檢測,對于低含量食品的檢測則受到儀器靈敏度的限制。另外,文獻報道的VB12的檢測方法還有原子吸收法[10]、毛細管電泳法[11-12]、酶聯免疫法[13-14]、電化學發光法[15]、電化學傳感器法[16]、液相色譜法[4,17-19]、液相色譜-串聯質譜法[20-23]、電感耦合等離子體質譜法[5,24]等,各個方法均存在一定的優缺點,且大多數方法主要針對部分基質進行方法的開發和應用。本研究主要考慮VB12作為一種在食品中應用較普遍的強化營養素,各種VB12強化配方食品及相關的保健食品均需檢測該指標,不必將嬰幼兒食品、乳品、保健食品及其他配方食品等基質分開建立檢測方法。而建立適合各類配方食品中VB12測定的通用分析方法,難點在于各類食品中VB12的含量范圍差異較大。因此本研究借助高效液相色譜-電感耦合等離子體-質譜(high performance liquid chromatography-inductively coupled plasma-mass spectrometry,HPLC-ICP-MS)聯用技術的高靈敏度與寬線性范圍優勢,建立適用于各種配方食品中VB12測定方法。由于VB12的不同結構形式的穩定性各不同,其中氰鈷胺素相較于其他幾種形態(羥鈷胺、腺苷鈷胺和甲基鈷胺等)更為穩定,配方食品中用于營養強化的VB12幾乎均為氰鈷胺素,因此,直接測定氰鈷胺素的含量,就能反映營養強化配方食品中VB12的添加水平,無需使用氰化鉀或氰化鈉等劇毒試劑進行結構轉化。本方法基于VB12結構中含有鈷元素,通過測定鈷的含量間接反映樣品中VB12(氰鈷胺素)的含量,與國家標準方法相比,本方法簡單、快速、靈敏度高、線性范圍廣,適用于各種配方食品中VB12的檢測,實驗結果較為滿意,在日常檢測方面存在一定的優勢。
1 材料與方法
1.1 材料與試劑
強化VB12的果汁飲料、谷物制品、含乳飲料、保健品 市售。
VB12(氰鈷胺素) 美國Sigma-Aldrich公司;乙酸銨(色譜純) 上海麥克林生化科技有限公司;乙腈、甲醇(均為色譜純) 德國Merck公司;甲酸(色譜純) 賽默飛世爾科技(中國)有限公司;鹽酸、三氯乙酸(均為分析純) 國藥集團化學試劑有限公司;所有用水均為超純水。
1.2 儀器與設備
DIONEX UltiMate 3000液相色譜儀、iCAP Q ICP-MS儀及工作站 美國賽默飛世爾儀器公司;Milli-Q去離子水發生器 美國Millipore公司;SB25-12DTS雙頻超聲波清洗器 廣州市鵬鑫科學儀器有限公司;ME204電子平梅特勒-托利多儀器(上海)有限公司;MS3 Digital渦旋儀德國IKA公司;Centrifuge 5810離心機 艾本德中國有限公司;0.22 μm水相濾膜 天津津騰公司;Oasis HLB柱(6 mL,500 mg) 美國Waters公司。
1.3 方法
1.3.1 HPLC條件
Thermo Fisher Hypersil GOLD C18色譜柱(100 mm×2.1 mm,3 μm);流動相:A為8 mmol/L乙酸銨溶液,B為甲醇(81∶19,V/V)溶液,等度洗脫,流速為0.4 mL/min;柱溫24 ℃;進樣量20 μL。
1.3.2 ICP-MS條件
選擇Co-59作為定量元素,采用KED模式,經調諧儀器主要參數如表1所示。

表1 ICP-MS條件Table 1 ICP-MS experimental conditions
1.3.3 樣品前處理
谷物制品:稱取試樣5 g(精確至0.01 g)于50 mL離心管中,加入25 mL 0.01 mol/L鹽酸溶液,渦旋混勻1 min,超聲15 min,3 900 r/min離心15 min。取上清液置于50 mL棕色容量瓶中,用水定容至刻度,待凈化。
含乳飲料:稱取試樣10 g(精確至0.01 g)于50 mL離心管中,加入10 mL 40 g/L三氯乙酸溶液,渦旋混勻1 min,超聲15 min,3 900 r/min離心15 min。取上清液置于50 mL棕色容量瓶中,用水定容至刻度,待凈化。
保健品:稱取試樣0.5 g(精確至0.01 g)于50 mL離心管中,加入25 mL 0.01 mol/L鹽酸溶液,渦旋混勻1 min,超聲15 min,3 900 r/min離心15 min。取上清液置于50 mL棕色容量瓶中,用水定容至刻度,待凈化。
1.3.4 樣品凈化
將待凈化液通過已活化的固相萃取柱(分別用5 mL甲醇和5 mL水預淋洗活化),用5 mL 7%乙腈溶液將干擾物質從固相萃取柱上淋洗下來,最后用3.0 mL 25%乙腈溶液將目標化合物洗脫,收集全部洗脫液,根據含量稀釋洗脫液,過0.22 μm微孔濾膜后,待檢測分析。
1.4 數據統計
采用Qtegra、MultiQuant 3.0.2軟件處理數據,采用Origin 8.0、Microsoft Excel 2010軟件繪圖。
2 結果與分析
2.1 方法的可行性
HPLC-ICP-MS法測定樣品中VB12的含量,主要是基于VB12結構中含有鈷元素,通過測定樣品中鈷的含量間接推算出VB12的含量。但樣品中的鈷除來源于VB12,也有可能來源于樣品中的含鈷雜質以及游離鈷,原子吸收法以及ICP-MS法只能對樣品中的總鈷進行測定,由于沒有進行有效分離,檢測結果較容易受到樣品中無機鈷元素的影響而造成測定結果比實際值高。
本實驗采用HPLC-ICP-MS法,首先對樣品進行HPLC分離,色譜柱選用C18柱,樣品中的游離鈷離子在C18色譜柱中不保留,從柱中很快洗脫下來,然后通過選擇合適的流動相,將樣品中的其他非VB12的含鈷雜質與VB12目標物有效分離,再進行ICP-MS測定。如圖1所示,在45、98 s和270 s均有鈷離子響應,根據VB12標準溶液的保留時間進行定性,270 s處的色譜峰即為目標鈷離子色譜峰,樣品經HPLC分離后可較好地去除樣品中的含鈷雜質及游離鈷對測定結果的干擾,并保證檢測數據的準確性,這與文獻[25]分析結果一致。
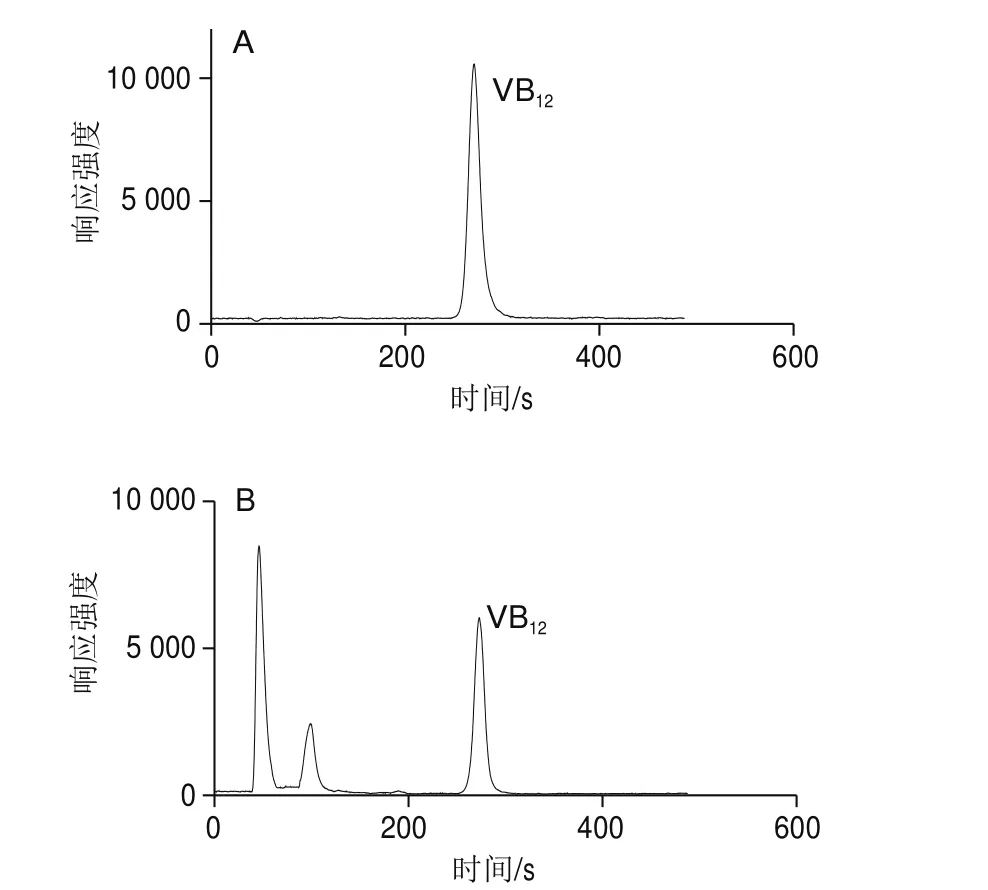
圖1 VB12標準溶液譜圖(10 μg/L)(A)與樣品溶液(B)譜圖Fig. 1 Chromatograms of VB12 standard solution (10 μg/L) (A) and sample solution (B)
2.2 HPLC條件的優化
本實驗選用C18色譜柱進行樣品分離,其流動相中通常會加入一定比例的有機溶劑以保護色譜柱,防止柱填料變性。但在ICP-MS中,有機溶劑的使用可能會導致采樣錐錐口處產生碳積,影響儀器的信號穩定性,高濃度的有機溶劑還會導致離子源熄火,影響實驗效率并增加儀器的維護成本。雖然有相關實驗表明[26],外加氧氣可以帶走碳,但同時也會導致儀器靈敏度下降。因此,本實驗嚴格控制流動相中有機相的比例,比較流動相中甲醇溶液體積分數分別為15%、19%、23%時,對VB12的分離效果的影響,得到對比色譜圖見圖2A。當流動相中甲醇溶液體積分數為19%時,VB12在270 s出峰,峰形對稱且尖銳;當流動相甲醇溶液體積分數調整為15%時,VB12在色譜柱中的保留性明顯增強,保留時間延長至750 s;增加流動相中甲醇溶液體積分數至23%時,VB12在150 s出峰,峰形保持對稱尖銳,且儀器的靈敏性沒有降低,碳滯留問題沒有出現。因此,在保證儀器靈敏度以及信號穩定性的前提下,應盡量選擇低有機相比例的流動相進行實驗,但綜合考慮分析效率,本實驗最終選擇19%甲醇溶液作為流動相中的有機相。
另外,比較水、8 mmol/L乙酸銨溶液、8 mmol/L乙酸銨溶液(pH 5.0)作為水相流動相時,對VB12分離效果的影響,見圖2B。水相流動相中引入8 mmol/L乙酸銨后VB12的峰形稍有改善,經測定,8 mmol/L乙酸銨溶液的pH值約為6.2。有研究報導[27],VB12在pH 4.5~5.0弱酸條件下最穩定,流動相pH值會改變VB12的色譜行為。但本實驗發現,將流動相pH值調至5.0時,VB12的響應和峰形幾乎沒有發生變化,通過48 h穩定性實驗,發現調節流動相pH值前后VB12在儀器上的響應沒有明顯變化,表明流動相pH值為6.2或5.0時,VB12的穩定性沒有明顯改變,因此,最終選擇以8 mmol/L的乙酸銨作為流動相的水相進行實驗。
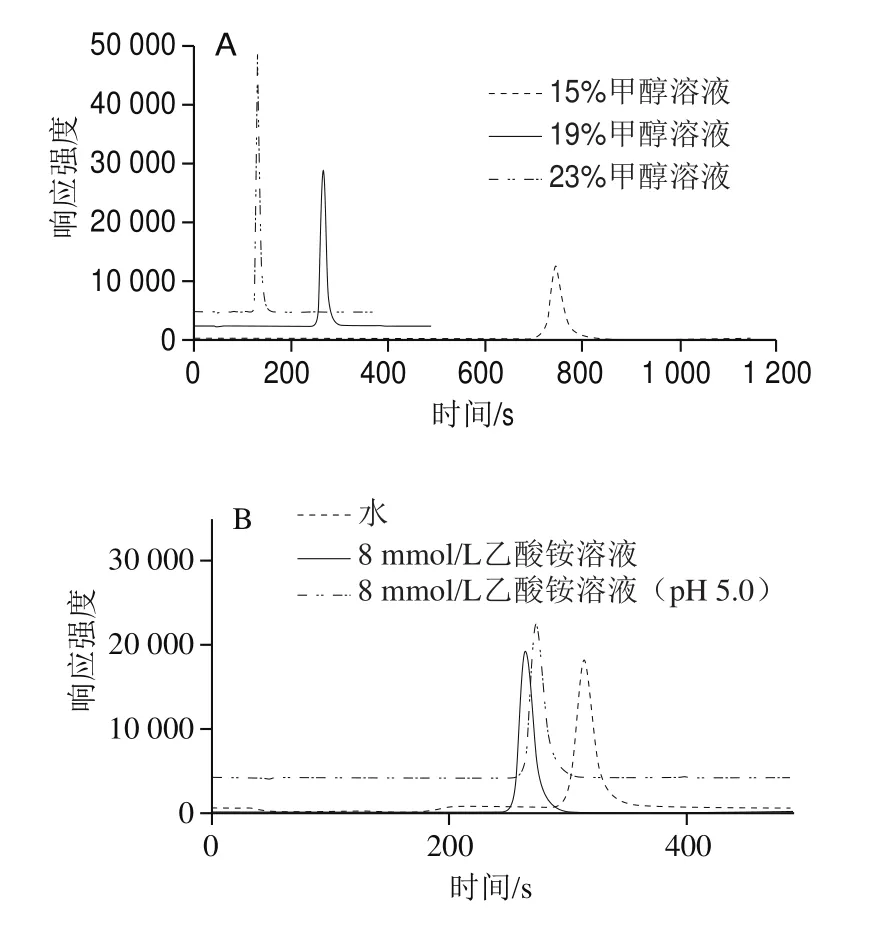
圖2 不同甲醇比例流動相(A)和水相流動相(B)對VB12分離效果的對比圖譜Fig. 2 Comparative separation of VB12 using different mobile phasesconsisting of methanol (A) and water phase (B)
2.3 樣品凈化的影響
由于食品基質復雜,研究者通常采用樹脂吸附法、透析法、免疫親和柱、固相萃取柱法等手段進行樣品凈化,其中固相萃取法以操作簡單、高效、可靠、消耗試劑少等優點備受青睞,在許多領域得到廣泛應用。本研究比較了實際樣品提取液經HLB固相萃取柱處理和不經HLB固相萃取柱處理的實驗結果,見表2,相應的對比圖譜見圖3。
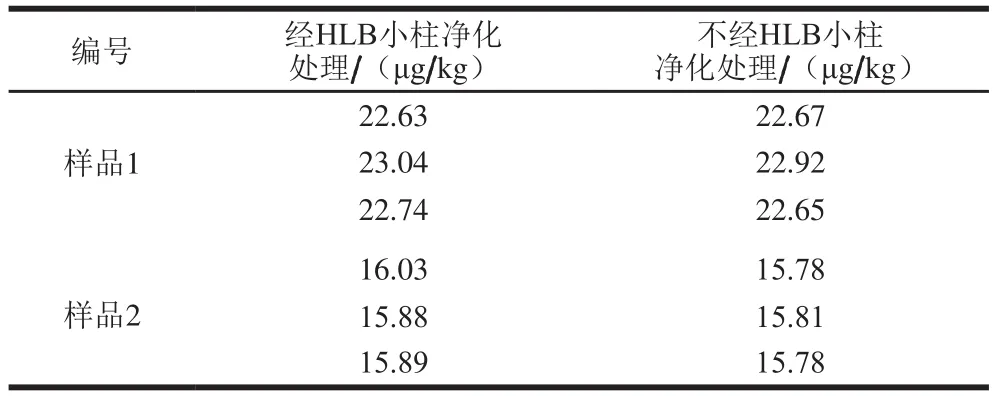
表2 凈化對HPLC-ICP-MS檢測結果的影響Table 2 Effect of puri fi cation on the results of HPLC-ICP-MS
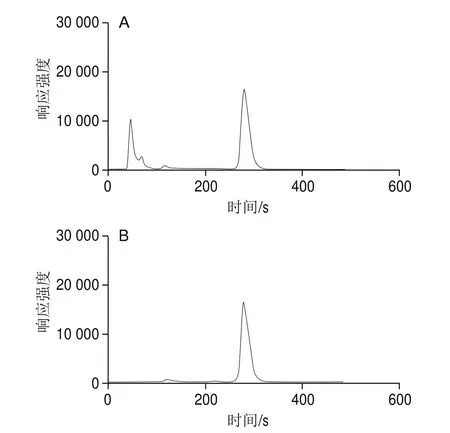
圖3 樣品凈化前(A)、后(B)對比圖譜Fig. 3 Comparative chromatogram of sample before (A) and after (B) purification
由表2可知,樣品提取液是否過柱對HPLC-ICP-MS測定VB12的結果幾乎無影響。圖3顯示,樣品提取液不過固相萃取柱也能在HPLC-ICP-MS上有較好響應,且與雜質能完全分離。但考慮到提取液過固相萃取柱后,能夠有效降低樣品中其他雜質的干擾,為減少雜質對色譜柱的損害作用,本實驗選擇采用HLB固相萃取小柱對提取液進行凈化。
2.4 方法驗證
2.4.1 線性關系和檢出限結果
配制一系列質量濃度的標準溶液在1.3節的儀器分析條件下進行測定,得出VB12在0.1~500 μg/L范圍內呈現良好的線性關系,回歸方程為y=13 549x+2 782,相關系數為0.999。采用空白樣品加標實驗確定檢出限,逐級稀釋至樣品溶液中目標峰響應為3 倍信噪比時確定VB12的檢出限為0.02 μg/kg。
2.4.2 回收率和精密度結果
選取不同基質、混合均勻的VB12陽性樣品,分別對樣品進行3 個水平的加標回收率實驗,每組實驗重復6 次,按照1.3.3節樣品前處理方法制備供試品溶液,按儀器條件進行分析,測定VB12的含量,并計算相應的回收率,結果見表3。果汁飲料回收率為96.0%~103.6%,相對標準偏差(relative standard deviation,RSD)為0.4%~2.8%;谷物制品回收率為81.3%~93.2%,RSD為1.4%~3.0%;含乳飲料回收率為84.0%~94.0%,RSD為0.9%~2.7%;保健品回收率為90.7%~103.3%,RSD為3.2%~4.6%,表明方法的準確度和重復性良好,能應用于不同基質的配方食品中VB12的檢測分析。

表3 食品中VB12的回收率和RSD(n= 6)Table 3 Recovery and repeatability (RSD) for VB12 from different spiked samples (n= 6)
2.4.3 儀器及標準溶液的穩定性
準確吸取標準品溶液,按儀器條件連續進樣6 次,測定峰面積,并計算RSD。得到標準溶液的峰面積RSD小于0.10%,保留時間RSD小于1.0%,表明儀器精密度良好。
準確吸取標準品溶液,在儀器條件下,分別在0、1、2、4、8、12、24、36、48 h進樣,以標準溶液的峰面積和保留時間為對象,計算RSD小于2.0%,表明標準溶液在48 h內穩定。
2.5 本方法與其他分析方法的比較
目前,已有檢測方法存在問題如下:微生物法靈敏度高,但操作步驟繁瑣、分析時間長,較為耗時;分光光度法、毛細管電泳法、HPLC法均不能用于食品中的微量或痕量VB12的測定分析;原子吸收光譜法,ICP-MS法測定VB12的原理也是通過測定鈷元素含量間接反映VB12含量,但這些方法容易受到樣品中無機鈷及其他含鈷雜質的影響而造成測定結果比實際值高;酶聯免疫法選擇性好、靈敏度高、實用性強,其檢測結果的準確性通常需要儀器分析法進一步驗證。研究表明,HPLC-MS/MS法測定VB12,操作簡單、靈敏度準確度高,方法可靠,是目前較為主流的分析手段。因此,將本實驗建立的HPLC-ICP-MS方法與HPLC-MS/MS法進行比較,通過重復測試同一實驗樣品的檢測結果進行對比分析。
將2.3節中處理的未經固相萃取小柱凈化和經固相萃取小柱凈化的樣品溶液于HPLC-MS/MS上進行檢測。從表4可以看出,樣品提取液是否凈化對HPLC-MS/MS的測定結果有較大影響,樣品1和樣品2不經HLB小柱凈化所測結果明顯低于經HLB小柱凈化后所測結果,表明樣品基質對于HPLC-MS/MS的檢測結果具有一定的影響。將表2與表4進行比較發現,樣品經HLB小柱凈化處理后,2 種檢測方法的測定結果基本一致,表明HPLC-ICP-MS方法的抗基質干擾能力要優于HPLC-MS/MS法。由于大多數產品中VB12的含量均較低,且食品基質相對復雜,對于此類樣品的分析,選擇抗基質干擾能力強的HPLC-ICP-MS方法進行測定具有一定的優勢,且目前HPLC與ICP-MS聯用技術已較為成熟,相關的應用報道[28-31]也較多,此聯用儀器也逐漸普遍化,可以采用此方法進行相關產品中VB12含量的分析檢測以及質量控制。
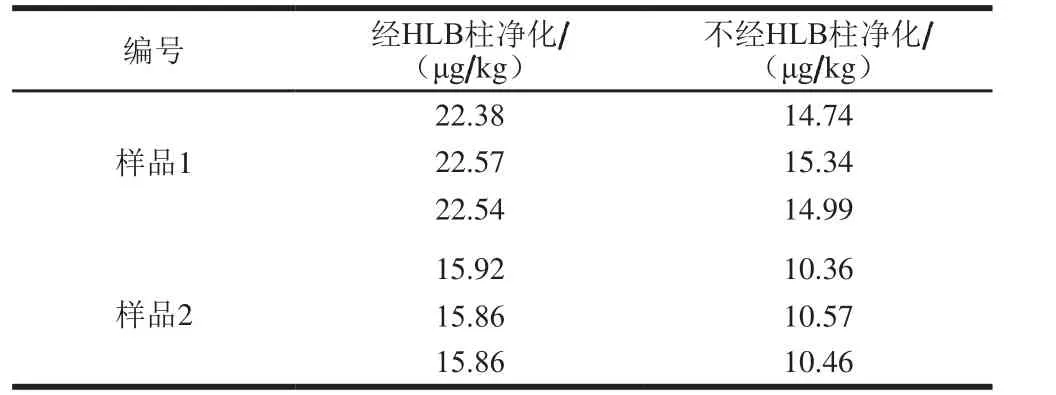
表4 凈化對HPLC-MS/MS檢測結果的影響Table 4 Effect of puri fi cation on the results of HPLC-MS/MS
2.6 實際樣品測定結果
應用建立的方法對購自超市的強化VB12的配方食品(果汁飲料、谷物制品、含乳飲料和保健品)進行檢測,結果表明不同果汁飲料中VB12的含量在0.52~1.14 μg/kg之間,部分標示為功能性飲料中的VB12含量高達15.83 μg/kg;谷物制品中VB12的含量在10.85~12.24 μg/kg之間;含乳飲料中VB12的含量在0.68~0.94 μg/kg之間;保健品中VB12的含量較高,測定值為3 049.15 μg/kg,折算后保健品中VB12的含量為4.57 μg/片。
3 結 論
本實驗針對不同基質的配方食品中VB12含量的測定方法進行研究,借助于HPLC-ICP-MS聯用技術的高靈敏度與寬線性范圍優勢,建立了適用于各種配方食品基質中VB12含量測定的HPLC-ICP-MS法。通過方法優化,得出VB12的方法檢出限為0.02 μg/kg,靈敏度高、峰形較好、分析時間短,可用于實際樣品的分析檢測。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
