分散固相萃取法結(jié)合UPLC-MS/MS同時(shí)分析茶葉中13種殺菌劑
2019-09-12 07:39:54劉文靜吳建鴻
福建茶葉 2019年5期
關(guān)鍵詞:檢測(cè)
黃 彪,劉文靜,吳建鴻
(福建省農(nóng)業(yè)科學(xué)院農(nóng)業(yè)質(zhì)量標(biāo)準(zhǔn)與檢測(cè)技術(shù)研究所/福建省農(nóng)產(chǎn)品質(zhì)量安全重點(diǎn)實(shí)驗(yàn)室,福建福州 350003)
1 前言
在農(nóng)業(yè)領(lǐng)域,殺菌劑主要是指用于防治由各種病原微生物引起的植物病害的一類(lèi)農(nóng)藥。殺菌劑能很好的防治植物病害,起到保護(hù)植物、促進(jìn)生產(chǎn)的作用,被廣泛應(yīng)用于農(nóng)作物的種植過(guò)程。由于農(nóng)藥殘留的潛在危害,許多國(guó)家及國(guó)際組織都對(duì)農(nóng)藥殘留嚴(yán)格規(guī)定了殘留限量[1-2]。我國(guó)茶葉品種資源多,種植區(qū)域廣,茶樹(shù)在種植過(guò)程中從葉、莖、根到花等均可能有病蟲(chóng)危害,因此殺菌劑在茶葉的種植過(guò)程中應(yīng)用較多,國(guó)際貿(mào)易中對(duì)農(nóng)產(chǎn)品中農(nóng)藥殘留最高限量的規(guī)定越來(lái)越嚴(yán)格,有關(guān)茶葉的農(nóng)藥殘留限量標(biāo)準(zhǔn)也更為嚴(yán)苛,而我國(guó)一直以來(lái)是重要的茶葉出口大國(guó),因此研究茶葉中殺菌劑殘留的檢測(cè)方法具有非常重要的意義。
快速、高效的農(nóng)藥殘留分析方法能否建立在很大程度上取決于樣品前處理過(guò)程,高效的前處理技術(shù)往往能大大減少分析的成本和時(shí)間。目前,農(nóng)產(chǎn)品中殺菌劑的檢測(cè)常見(jiàn)前處理方法有液液萃取、固相柱萃取、分子印跡固相萃取及分散固相萃取等[3-7]。分散固相萃取技術(shù)相比于其他前處理方法具有溶劑用量少,提取速度快,重現(xiàn)性好等優(yōu)點(diǎn),在樣品前處理過(guò)程中得到廣泛應(yīng)用。殺菌劑的檢測(cè)方法主要有毛細(xì)管電泳法[8]、氣相色譜法[9-10]、液相色譜法[11-12]、氣相色譜-串聯(lián)質(zhì)譜法[13-14]、液相色譜-串聯(lián)質(zhì)譜法[15-17]等。
質(zhì)譜檢測(cè)技術(shù)因其抗干擾強(qiáng),檢測(cè)快速,靈敏度高,定性定量準(zhǔn)確等優(yōu)點(diǎn),已逐漸發(fā)展成為分析檢測(cè)中的主導(dǎo)技術(shù),而尤其是超高效液相色譜-串聯(lián)質(zhì)譜分析技術(shù),將超高效液相色譜儀對(duì)復(fù)雜樣品的快速分離能力與質(zhì)譜儀對(duì)目標(biāo)離子的高選擇性、高靈敏度相結(jié)合,在食品安全、藥物分析、環(huán)境監(jiān)測(cè)等領(lǐng)域中越來(lái)越發(fā)揮出重要作用。在已有報(bào)道中,利用分散固相萃取結(jié)合超高效液相色譜-串聯(lián)質(zhì)譜法檢測(cè)茶葉中殺菌劑殘留的報(bào)道尚不多見(jiàn)。本研究利用分散固相萃取法前處理凈化,建立了超液相色譜-串聯(lián)質(zhì)譜法分析茶葉中殺菌劑殘留。該方法樣品前處理時(shí)間短,分析快速,準(zhǔn)確性好,可為茶葉殺菌劑殘留定性定量分析提供方法參考依據(jù)。
2 材料與方法
2.1 儀器與試劑
ACQUITY UPLC H-Class超高效液相色譜儀(美國(guó)Waters公司);Xevo TQ-S三重四極桿質(zhì)譜儀(美國(guó)Waters公司);樣品均質(zhì)器(德國(guó)IKA公司);SYG-2水浴恒溫振蕩器(常州朗越儀器有限公司);XW-80A旋渦混合器(上海醫(yī)科大學(xué)儀器廠);Millipore Qirect-Q5超純水設(shè)備(美國(guó)Millipore公司)。
乙腈(色譜純,美國(guó)Waters公司)、甲醇(色譜純,美國(guó)Waters公司)、乙酸銨(LC-MS級(jí),美國(guó) Waters公司)、甲酸(LC-MS級(jí),美國(guó)Waters公司)、N-丙基乙二胺(PSA,40 μm,天津博納艾杰爾科技有限公司)、C18吸附劑(40-60 μm,天津博納艾杰爾有限公司)、超純水(Millipore超純水機(jī)制備),其他試劑為分析純。
2.2 標(biāo)準(zhǔn)溶液的配制
氟硅唑、丙環(huán)唑標(biāo)準(zhǔn)品(純度97%,德國(guó)DR公司),用乙腈溶解配制成濃度為1 000 mg/L的標(biāo)準(zhǔn)儲(chǔ)備液;三唑酮、苯醚甲環(huán)唑、多效唑等農(nóng)藥標(biāo)準(zhǔn)液購(gòu)于國(guó)家標(biāo)準(zhǔn)物質(zhì)中心,濃度規(guī)格為1 000 mg/L。標(biāo)準(zhǔn)品儲(chǔ)備液置于-20℃冰箱中保存,實(shí)驗(yàn)時(shí)取儲(chǔ)備液配制成10 mg/L的單標(biāo)中間液,用空白基質(zhì)溶液配制的用于制作標(biāo)準(zhǔn)曲線的不同濃度的混標(biāo)工作液,現(xiàn)配現(xiàn)用。
2.3 樣品前處理方法
準(zhǔn)確稱(chēng)取5.00 g(精確至0.01 g)磨碎的茶葉樣品于50 mL離心管中,加入10 mL乙腈,用均質(zhì)器以15000 r/min均質(zhì)提取1 min后,蓋好蓋子振蕩提取15min,以 8 000 r/min離心5 min;轉(zhuǎn)移上清液2.0 mL到預(yù)先加有50 mg PSA,50 mg C18,10 mg石墨化炭黑GCB的10 mL離心管中,渦旋30 s后,8 000 r/min離心1min;取上清液過(guò)0.22 μm 濾膜,濾液供UPLC-MS/MS分析。
2.4 色譜-質(zhì)譜條件
超高效液相色譜條件:Waters T3 C18色譜柱(2.1 mm×50 mm,1.8 μm,);柱溫:30.0℃;梯度洗脫:流動(dòng)相A為0.1%甲酸-5 mmol/L乙酸銨水溶液,流動(dòng)相 B 為乙腈;0~3 min,90%~70%A ;3~5 min,維持 70%A;5~8 min,70%~20%A;8~10 min,20%~90%A;流動(dòng)相流速:0.2 mL/min;進(jìn)樣量:4 μL。
質(zhì)譜條件:電噴霧ESI+掃描方式;檢測(cè)方式:多反應(yīng)監(jiān)測(cè)(MRM)模式;毛細(xì)管電壓:2.5 kV;錐孔氣流:氮?dú)猓魉?0 L/h;脫溶劑溫度:150℃,流速 800 L/h;離子源溫度:150 ℃;霧化氣壓力:0.28 MPa;碰撞氣:高純氬氣。相關(guān)質(zhì)譜檢測(cè)參數(shù)見(jiàn)表1。
3 結(jié)果與討論
3.1 色譜-質(zhì)譜條件的優(yōu)化
在實(shí)驗(yàn)過(guò)程中,比較了以甲醇-水和乙腈-水作為流動(dòng)相以及在流動(dòng)相中添加甲酸、乙酸銨對(duì)目標(biāo)化合物的分離效果及目標(biāo)離子色譜峰峰型的影響。實(shí)驗(yàn)結(jié)果表明:采用乙腈-5 mmol/L乙酸銨水溶液(含0.1%甲酸)作為流動(dòng)相時(shí),在正離子掃描模式下,殺菌劑有良好的峰形且相對(duì)于不加甲酸的流動(dòng)相有更好的響應(yīng)強(qiáng)度,甲酸的加入有利于目標(biāo)分子更好的形成正離子,5 mmol/L乙酸銨-0.1%甲酸水溶液緩沖體系在一定程度上可以調(diào)節(jié)體系的pH值,使得在一個(gè)合適的酸堿性范圍內(nèi)目標(biāo)離子有更好的峰型。
3.2 提取溶劑的選擇
在農(nóng)藥目標(biāo)分子的前處理過(guò)程中,常用的萃取溶劑有甲醇、乙腈、乙酸乙酯等。甲醇雖對(duì)農(nóng)藥分子有很好的溶解性,但甲醇是一種質(zhì)子性溶劑在提取農(nóng)藥分子的同時(shí)也很容易將樣品中的醇類(lèi)、多酚、有機(jī)酸等物質(zhì)提取出來(lái)。茶葉樣品中既有較多的茶多酚等物質(zhì),同時(shí)也含有沒(méi)食子酸等一些有機(jī)酸物質(zhì),采用甲醇作為提取茶葉樣品中的農(nóng)藥分子,會(huì)對(duì)后續(xù)的凈化過(guò)程造成一定的影響。乙睛作為一種極性較大的溶劑、穿透力強(qiáng),對(duì)農(nóng)藥分子的提取效率高,并且用乙腈作為提取溶劑可以減少維生素、色素等中等極性雜質(zhì)的溶出,因此被廣泛用于殘留分析中目標(biāo)分子的提取。如用乙酸乙酯作為提取溶劑,最后需氮吹轉(zhuǎn)溶劑為乙腈或者甲醇進(jìn)行液相色譜-質(zhì)譜分析,會(huì)使得前處理時(shí)間變長(zhǎng)。因此綜合考慮,采取乙腈作為提取溶劑。

表1 13種殺菌劑檢測(cè)的質(zhì)譜參數(shù)
3.3 分散固相萃取劑的選擇
在樣品前處理過(guò)程中常用的固相萃取劑有PSA、C18及GCB。PSA是含有氨基基團(tuán)的強(qiáng)極性分子,能夠通過(guò)強(qiáng)的吸附及離子交換過(guò)程將與基質(zhì)中的多酚、有機(jī)酸、糖類(lèi)等含有羥基或者羧基的極性分子除去。C18由于分子中鍵合十八烷基,對(duì)極性較小的分子具有良好的吸附作用,能夠去除基質(zhì)中的色素、維生素等物質(zhì)。GCB則對(duì)色素有良好的去除能力。考慮到茶葉基質(zhì)中有茶多酚、有機(jī)酸、維生素及色素等物質(zhì),因此在實(shí)驗(yàn)中考慮將上述吸附劑進(jìn)行組合對(duì)樣品前處理凈化。在實(shí)驗(yàn)中以茶葉樣品為基質(zhì)加入50 μg/kg混標(biāo)溶液,考察了不同用量固相萃取劑的凈化效果及回收率情況:(1)25 mg PSA+25 mg C18+10 mg GCB(2)50 mg PSA+50 mg C18+10 mg GCB(3)75 mg PSA+75 mg C18+10 mg GCB。實(shí)驗(yàn)結(jié)果表明:以乙睛為提取溶劑的情況下,第(2)、(3)兩種吸附劑綜合使用效果都較好,能夠較好的對(duì)樣品前處理去除雜質(zhì),并且目標(biāo)分子的回收率均在75%以上。另外也考察了PSA和C18用量不變時(shí),改變GCB用量?jī)艋Ч牟煌海?)50 mg PSA+50 mg C18+5 mg GCB (5)50 mg PSA+50 mg C18+10 mg GCB(6)50 mg PSA+50 mg C18+15 mg GCB。實(shí)驗(yàn)中發(fā)現(xiàn),加入 10 mg GCB即可很好的去除色素等雜質(zhì)。因此,綜合考慮實(shí)驗(yàn)成本、凈化效以及對(duì)回收率的影響,實(shí)驗(yàn)中采用50 mg PSA+50 mg C18+10 mg GCB吸附劑組合進(jìn)行樣品的凈化處理。
3.4 方法評(píng)價(jià)
將空白樣品經(jīng)固相萃取劑分散提取得到基質(zhì)凈化液,配制13種殺菌劑的基質(zhì)匹配混標(biāo)溶液,注入超高效液相色譜-串聯(lián)質(zhì)譜儀進(jìn)行分析。以目標(biāo)物特征離子色譜峰響應(yīng)信號(hào)峰面積(y)為縱坐標(biāo),目標(biāo)化合物的基質(zhì)匹配溶液濃度(μg/L)x為橫坐標(biāo),制作標(biāo)準(zhǔn)曲線。依據(jù)特征離子色譜峰信號(hào)的3倍信噪比(S/N≥3)確定方法的檢出限,根據(jù)各目標(biāo)化合物的響應(yīng)情況在茶葉基質(zhì)中進(jìn)行低、中、高3個(gè)濃度水平的加標(biāo)。結(jié)果表明:13殺菌在0.5 μg/L~100 μg/L濃度范圍內(nèi)基質(zhì)匹配曲線線性關(guān)系良好(r≥0.995),方法靈敏度高,目標(biāo)化合物的檢出下限在 0.08~0.7 μg/kg之間。不同濃度水平加標(biāo)相應(yīng)平均回收率為70.1%~106.7%,相對(duì)標(biāo)準(zhǔn)偏差在 1.2%~7.6%之間(表 2)。
4 實(shí)際樣品的檢測(cè)
采集茶葉樣品40余份進(jìn)行了殺菌劑殘留的分析,發(fā)現(xiàn)1份茶葉樣品中有苯醚甲環(huán)唑殘留,含量為0.056 mg/kg,陽(yáng)性樣品的MRM離子色譜圖如圖1所示。雖有檢出,但未超出食品安全國(guó)家標(biāo)準(zhǔn)限量規(guī)定(標(biāo)準(zhǔn)規(guī)定茶葉中苯醚甲環(huán)唑最大殘留限量為10 mg/kg)[2]。
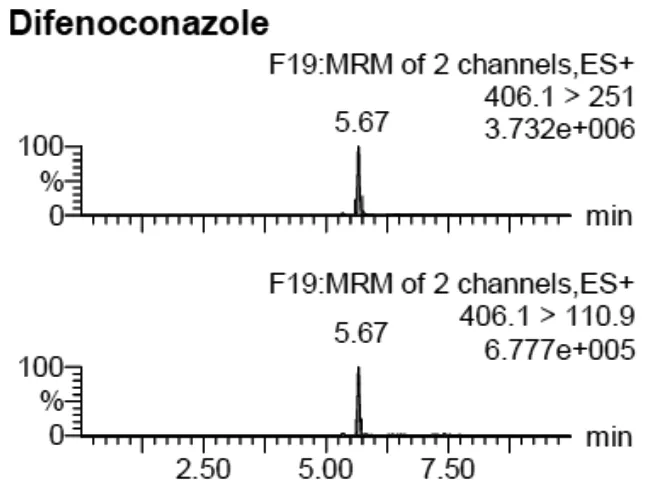
圖1 陽(yáng)性樣品的苯醚甲環(huán)唑MRM離子色譜圖

表2 13種殺菌劑的平均加標(biāo)回收率和檢出限(n=6)
5 結(jié)論
本研究運(yùn)用分散固相萃取前處理法結(jié)合超高效液相色譜-串聯(lián)質(zhì)譜法,建立了茶葉中多菌靈、三唑酮、腈菌唑、苯醚甲環(huán)唑等13種殺菌劑殘留的檢測(cè)方法。該方法前處理簡(jiǎn)單,方便快速,具有良好的靈敏度、準(zhǔn)確度和精密度,可用于茶葉中殺菌劑殘留的同時(shí)分析,有利于農(nóng)產(chǎn)品質(zhì)量安全的保障。
猜你喜歡
中國(guó)設(shè)備工程(2022年12期)2022-07-11 04:33:00
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2019年9期)2019-11-25 07:34:36
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2019年9期)2019-11-25 07:34:34
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2019年12期)2019-05-21 02:53:50
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2019年12期)2019-05-21 02:53:48
