hcp-Co基費托合成催化劑的研究進展
2019-10-11 03:06:50寧文生
石油化工 2019年9期
關鍵詞:催化劑
王 凱,代 惠,李 貝,王 彪,寧文生
(浙江工業大學 化學工程學院,浙江 杭州 310014)
費托(F-T)合成反應可將非石油基的各類含碳資源,如煤、生物質和天然氣等經合成氣(CO+H2)加氫轉變為清潔液體燃料和基礎化工原料,不僅可緩解石油資源的緊缺,而且產物對環境友好(無硫、無氮、無芳烴),還可避免煤炭等化石燃料直接燃燒造成的環境污染,因此F-T 合成技術的合理利用,對于改善環境污染、緩解石油資源的短缺都具有重要的現實意義。Fe和Co催化劑兼具優異的F-T合成催化性能和合理的成本優勢,一直受到學術界和工業界的廣泛關注。與Fe 基催化劑相比,Co 基催化劑具有高活性、低CH4選擇性、低水煤氣變換活性和穩定性好等優點,是富含H2的合成氣生產中間餾分和重質蠟的最佳選擇[1]。
本文從Co 基催化劑活性中心的晶型入手,簡要歸納和總結了F-T 合成催化劑的制備方法和理論計算的研究進展。
1 Co 基催化劑的反應活性結構
目前,對Co 基催化劑的活性中心形成了較為一致的認知,即0 價的金屬Co 是F-T 合成的活性相。金屬Co 主要以六方密堆積(hcp)相和面心立方(fcc)相兩種晶型存在,hcp-Co 在較低溫度下是穩定的,但在400 ℃左右hcp-Co 會發生相變向fcc-Co 轉換[2]。Garces 等[3]利用原位XRD 方法研究了無載體的Co3O4在H2氣氛中的還原過程,發現在250~300 ℃時,Co3O4先被還原成CoO,再被還原成hcp-Co,還原溫度高于400 ℃的條件下,僅獲得fcc-Co,但當分步進行還原時,在450℃條件下得到了hcp-Co 和fcc-Co 的混合物。
大量研究結果表明,在F-T 合成反應過程中,Co 基催化劑中存在hcp-Co 和fcc-Co 兩種晶型,且hcp-Co 的F-T 合成催化活性高于fcc-Co。Liu等[4]針對F-T 合成中Co 活性相的最佳尺寸和晶體結構等問題,通過核磁共振波譜(NMR)和溫差核磁共振波譜(TDFNR)研究了催化劑中Co 納米顆粒的尺寸、結構和原子空間排列順序,Co/SiC和Co/TiO2-SiC 催化劑的活性相主要為hcp-Co,催化活性隨著Co 納米顆粒暴露的總比表面積的增加而提高;而在Co/CNT-Al2O3催化劑中活性相為fcc-Co,無論Co 納米顆粒暴露的總比表面積增大還是減小,Co/CNT-Al2O3的催化性能都低于Co/SiC 和Co/TiO2-SiC 催化劑,證明了hcp-Co 的形成是催化劑F-T 合成活性提高的根本原因。Jung等[5]采用浸漬法制備了Ru-Co/γ-Al2O3催化劑,并進行TPR 和XRD 等表征。實驗結果表明,Ru 的添加導致Co3O4的還原峰向低溫移動;在H2氣氛中焙燒Ru-Co/γ-Al2O3催化劑,還原后部分金屬Co以hcp晶型存在,而在空氣氣氛中焙燒的催化劑,還原后金屬Co 只有fcc 結構;不同氣氛焙燒后的催化劑,均在450 ℃條件下用H2/N2混合氣還原16 h 后進行F-T 合成反應性能評價,結果發現H2氣氛焙燒的催化劑對F-T 合成顯示出較高的催化活性和選擇性。Mitchel 等[6]采用浸漬法制備了以SiO2,Al2O3,TiO2,ZrO2等為載體的4 種Co 基催化劑,考察了焙燒操作條件對催化劑結構和性能的影響。相對于焙燒后再還原,Co/ZrO2未經焙燒而直接還原可產生更多的hcp-Co,表征結果顯示吸附在hcp-Co 上的CO 比吸附在fcc-Co 上的CO 更容易發生CO 解離,這導致了催化劑活性更高。
研究者們通過理論計算發現,hcp-Co 和fcc-Co 活性和選擇性的巨大差異歸因于它們不同的表面結構和暴露晶面。Liu 等[7]基于密度泛函理論(DFT)的動力學計算,比較了hcp-Co 和fcc-Co上的CO 活化規律,發現在CO 活化過程中,hcp-Co 不僅具有比fcc-Co 更高的內在活性,而且具有不同的活化途徑,即在hcp-Co 表面傾向于直接解離CO,而不是像在fcc-Co 上的H 輔助解離CO,即使考慮到H 的存在,hcp-Co 仍然比fcc-Co 更有利于CO 的解離活化;由于hcp-Co 的晶體結構對稱性相對較低,能夠暴露出大量的高活性晶面,因此hcp-Co 具有更高的本征活性;并且CO 的吸附和解離能夠顯著影響催化劑活性,CO 存在橋式吸附和線式吸附兩種吸附類型,橋式吸附的CO 傾向于直接解離,因而更具活性[8-10]。研究者們在實驗中發現,hcp-Co 上的CO 橋式吸附量和解離能力都高于fcc-Co,這可能是導致hcp-Co 的F-T 合成催化活性高于fcc-Co 的原因。
2 hcp-Co 基催化劑的制備方法
Srinivasan 等[11]通 過 原 位XRD 表 征 負 載 在SiO2載體上的Co 基催化劑,發現在350 ℃條件下用H2還原的催化劑中同時存在hcp-Co 和fcc-Co,兩者的相對比例約為3∶7,并且發現約17%(w)的hcp 晶面上存在缺陷。Co 顆粒大于40 nm 時,hcp-Co 是主要晶型,小于20 nm 時,fcc-Co 是主要晶型[12];而van Dillen 等[13-14]的研究表明,6~8 nm 的Co 顆粒對F-T 合成反應最有利。為了制備出小于10 nm 的hcp-Co 顆粒,在載體類型、還原條件、合成方法、助劑、活化過程和Co 前體種類等方面對hcp-Co 晶型形成的影響進行研究。
2.1 載體類型和載體改性的影響
Enache 等[15]對ZrO2和Al2O3載體負載的Co基催化劑進行原位XRD 表征,發現催化劑焙燒后產生的Co3O4經還原會促進fcc-Co 的形成,但直接還原未焙燒的Co/ZrO2可以促進無定形或結晶度低的hcp-Co 的形成。Ghampson 等[16]采用具有不同孔徑的SiO2制備了負載Co 催化劑,運用XRD分析新鮮催化劑、還原后催化劑和反應后催化劑,發現還原后催化劑中存在fcc-Co 和hcp-Co。fcc-Co 的粒徑隨著載體孔徑的減小而變小,hcp-Co 的粒徑始終小于fcc-Co 的粒徑,且hcp-Co 的粒徑在反應前后保持不變。Andreev 等[17]利用NMR 技術揭示載體晶型對金屬Co 結構的影響,負載在兩種Al2O3載體上的Co還原后出現不同的晶體結構,Co/δ-Al2O3催化劑中的Co 均 為fcc-Co,而Co/γ-Al2O3催化劑中的Co 為hcp-Co 和fcc-Co 的混晶,并有較大的Co 納米顆粒(約70 nm)分布在載體表面上。Karaca 等[18-19]的研究表明,Pt-Co/Al2O3催化劑經H2還原后,同時出現了hcp-Co 和fcc-Co 兩種晶型(摩爾比約為1∶4),在F-T 合成反應中,hcp-Co 和fcc-Co 的摩爾比保持不變。黃健[20]采用不同含量的Nb 對SiO2載體進行改性,再用來制備負載型Co 基催化劑,Nb2O5-SiO2界面的存在能夠降低Co3O4顆粒與SiO2之間的相互作用,使Co3O4更容易被還原成金屬Co,促進hcp-Co 的形成,表征結果顯示,SiO2中Nb 改性量為1.57%(w)時,催化劑的F-T 反應活性最高。Liu等[21]在Al2O3表面上預涂CoAl2O4獲得改性Al2O3載體,然后浸漬法獲得Co 基催化劑。H2-TPR 表征結果顯示,催化劑的還原溫度隨CoAl2O4改性量的增加而降低,表明Co 離子與改性后的Al2O3載體之間的相互作用逐漸變弱;原位XRD 表征結果顯示,改性Al2O3載體負載的催化劑有高比例的hcp-Co 存在,而未經改性Al2O3負載的催化劑還原后只有fcc-Co。Wang 等[22]發現在230~235 ℃,2 MPa,V(H2)∶V(CO)=2 的條件下,催化劑的反應活性與載體中CoAl2O4含量呈火山形依賴關系,在Co/Al 摩爾比為0.08 時達到最大值,催化劑活性主要由hcp-Co 的生成和還原度與分散度之間的協同效應兩個因素決定。
2.2 助劑和還原氣氛的影響
邱成武等[23]采用浸漬法制備了不同Ru 添加量的Co/SiO2催化劑,在400 ℃下用H2還原,XRD表征結果顯示,加入Ru 后催化劑未檢測出Co 的氧化物,且當Ru 添加量為0.5%(w)時,金屬Co 主要以hcp-Co 形式存在。O’Shea 等[24-26]研究了還原氣氛(H2,CO,H2/CO)對Co/SiO2催化劑結構和F-T 合成反應性能的影響,采用H2/CO 合成氣還原催化劑可使CO轉化率大幅提高至90%(比H2還原的催化劑活性高5 倍);H2/CO 合成氣還原得到均勻分散的hcp-Co 納米顆粒,并有羰基鈷物種形成(H2還原得到fcc-Co 且Co 顆粒大小不一),這可能是由于合成氣還原出的hcp-Co 顆粒表面被碳納米纖維封裝,導致表面自由能降低并抑制相變。穆仕芳等[27]利用XRD,EDS,TEM,Raman,TG,H2-TPR 等方法表征了一次還原氣氛(H2、CO、H2/CO、先H2后CO、先CO 后H2)對Co/SiO2催化劑結構的影響,并考察了再經H2二次還原后催化劑的F-T 合成反應性能。一次還原氣氛為H2或先H2后CO 時,催化劑的活性相主要是fcc-Co,后者被檢測出嚴重的積碳;一次還原氣氛為CO、先CO 后H2或H2/CO 時,催化劑中出現fcc-Co 和hcp-Co 的混晶,前兩者都被檢測出有積碳生成。F-T 合成反應結果表明,一次還原氣氛分別為H2、CO 和H2/CO 合成氣時,催化劑活性相近;一次還原氣氛為先CO 后H2時,CO 轉化率最高,這是因為先CO 后H2的還原方式生成了大量有利于提高F-T 合成反應活性的hcp-Co,且H2還原消除了部分積碳。
2.3 催化劑的還原-碳化-還原活化
Ducreux 等[28]發 現Co2C 中 的Co 呈 現 出 與金屬相似的六邊形排列,且堆積順序與hcp 晶型(ABABAB)相同,可在約200 ℃下通過H2還原轉化為主要為hcp 晶型的金屬Co。由此他們提出了典型的還原-碳化-還原(RCR)活化方法,獲得具有高活性的hcp-Co 基催化劑。首先將催化劑中的Co 用H2還原為fcc-Co,隨后將還原出的fcc-Co 用CO 碳化形成Co2C,再用H2還原Co2C得到hcp-Co 基催化劑。在RCR 活化方法中,在常壓下用CO 碳化fcc-Co 速度很慢,在230 ℃、常壓條件下用CO 碳化還原后的Co/Al2O3催化劑16 h 后仍然存在fcc-Co 的衍射峰[29],說明碳化過程未完全完成。Kwak 等[30-31]的研究表明fcc-Co向Co2C 的轉化速度取決于反應壓力,220 ℃、2 MPa 為Pt-Co/γ-Al2O3催化劑的最佳碳化條件,從XRD 表征結果中可以看出全部的Co 都被碳化為Co2C,在隨后的H2還原過程中,Co2C 的脫碳開始于160 ℃左右,同時形成輕質烴,還原后的Co 均為hcp-Co;他們還發現對失活的Co 基催化劑通過簡單的碳化和還原步驟可重新獲得催化活性,甚至超過初始的催化活性。Patanou 等[32]將RCR 活化方法應用于Re-Co/γ-Al2O3催化劑,TG 和Raman表征結果顯示所有碳化試樣都含有大量積碳,但最終H2還原步驟可除去積碳,推測積碳的除去是影響催化劑性能的關鍵因素。Gnanamani 等[33]對Co/SiO2催化劑采用RCR 法活化得到的hcp-Co 和常規H2還原得到的fcc-Co 晶粒大小分別為20.4 nm和26.9 nm,說明hcp-Co 在催化劑中的分散度高于fcc-Co。Tsakoumis 等[34]采 用RCR 法 活 化Re-Co/γ-Al2O3,先在350 ℃、常壓、H2氣氛中還原10 h,再在230 ℃、1.4 MPa 條件下用CO 碳化3 h,最后分別在200,250,350,450 ℃的不同溫度下進行二次H2還原,都有hcp-Co 生成;相對于直接用H2還原的催化劑,通過RCR 法活化的催化劑反應活性都會增高,且各催化劑活性隨著二次H2還原溫度升高呈現出火山形分布,350 ℃還原能夠賦予催化劑最佳的反應活性和選擇性;二次H2還原溫度小于250 ℃時,催化劑中殘留大量積碳,導致催化劑活性的降低以及CH4選擇性的提高,而二次還原溫度為450 ℃時,還原出的hcp-Co 會發生部分相變,變為fcc-Co,從而導致催化劑活性的降低;催化劑的活化過程及評價結果見圖1。
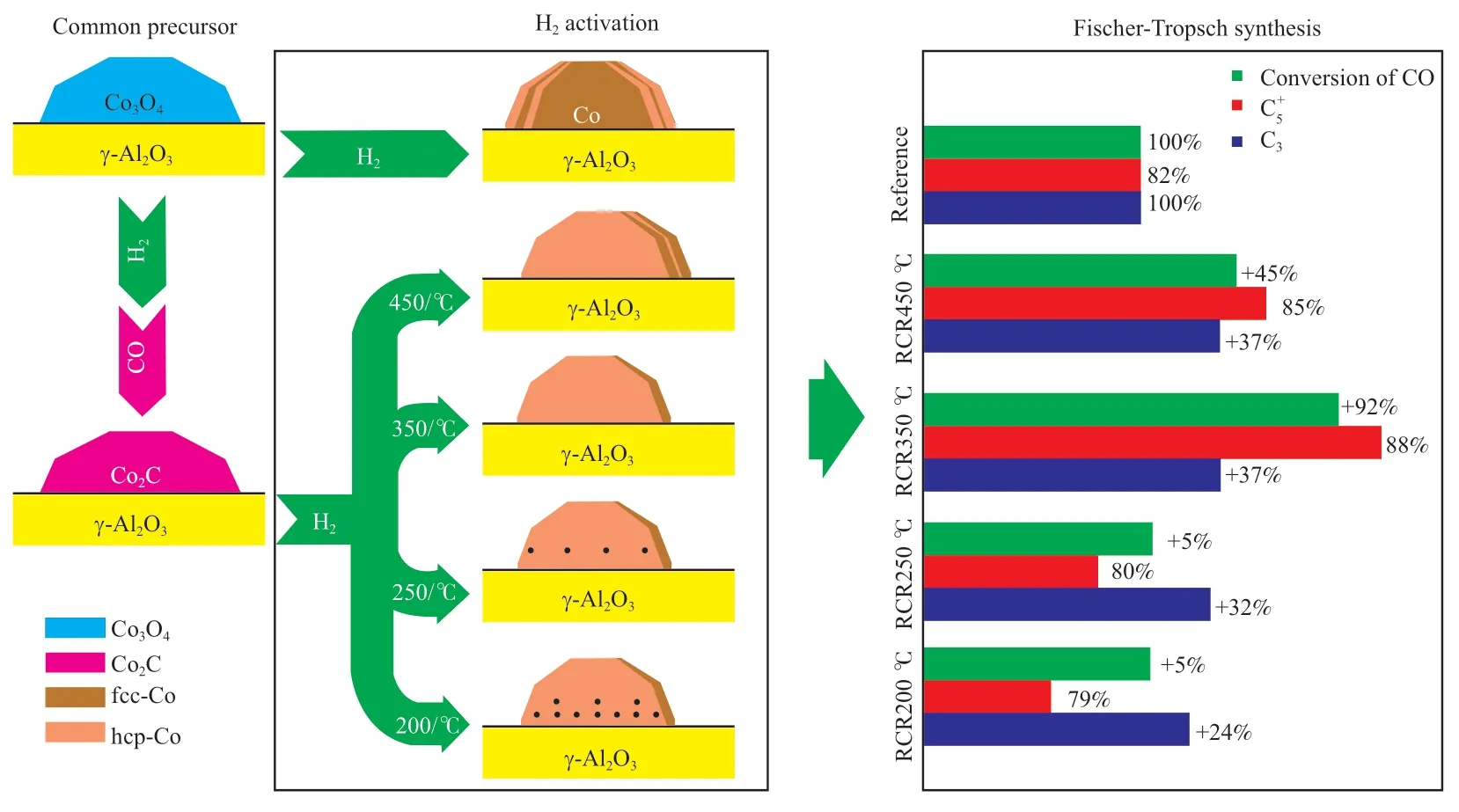
圖1 Re-Co/γ-Al2O3催化劑的不同活化過程及評價結果[34]Fig.1 Different activation processes and evaluation results of Re-Co/γ-Al2O3catalyst[34].
2.4 新型制備方法
2.4.1 還原CoO 前體
CoO 有具有八面體Co2+離子的立方巖鹽CoO(c-CoO,空間群Fm3m)結構和具有四面體Co2+離子的六方纖鋅礦CoO(h-CoO,空間群P63mc)結構兩種結晶相[35]。Seo 等[36]通過將Co(acac)3置于油胺中、在Ar 氣氛下加熱至135 ℃下溶解,然后立即通過快速加熱至200 ℃引發反應,退火1 h 后,將生成物冷卻至室溫,通過離心分離并用無水乙醇洗滌純化可得到綠色的h-CoO 納米晶體;在相似的合成條件下于135 ℃下反應更長的時間可得到棕色c-CoO 納米晶體;同時發現通過改變前體濃度和反應溫度,可很好地控制CoO 納米晶體的形狀和尺寸。Lü 等[37]在上述研究的基礎上,發現用H2還原h-CoO 可以選擇性的得到hcp-Co,用H2還原c-CoO 可選擇性的得到fcc-Co,且hcp-Co 和fcc-Co 的粒徑基本相同(小于 100 nm)。
2.4.2 還原Co{N(SiMe3)2}2(thf)前體
Wetz 等[38-39]在月桂酸(LA)和十六烷基胺(HDA)存在下,通過H2還原[Co{N(SiMe3)2}2(thf)]來制備hcp-Co 納米棒(NR)并選擇性暴露(11-20)晶面,由于金屬Co 是F-T 合成的活性相,這些NR 可直接用作F-T 合成催化劑而無需還原。Soulantica 等[40]在此基礎上運用種子誘導生長的方法,即用浸漬法制備的Co/SiO2-Al2O3中的Co顆粒當作種子,在LA 和HDA 存在下,用H2還原[Co{N(SiMe3)2}2(thf)]中的Co2+,并使其在種子表面外延生長,XRD 表征結果顯示,通過種子誘導生長的Co 表現出hcp-Co 晶型特征;評價結果表明,相比于常規浸漬法制備的催化劑,采用該方法制備的催化劑表現出更優異的活性和穩定性。Katerina 等[41]通過溶液外延生長法,使H2還原[Co{N(SiMe3)2}2(thf)]得 到 的hcp-Co 納 米 線在Cu 泡沫載體表面外延生長,SEM 表征結果顯示hcp-Co 納米線長度約1 μm、直徑約15 nm。由于采用Cu 泡沫作為載體,能夠更好地傳遞反應熱,從而限制了CH4形成并有利于產物的碳鏈增長。
2.5 Co 納米顆粒的制備方法
Wen 等[42]以CoSO4為Co 源、水合肼為還原劑、CTAB 為表面活性劑、檸檬酸鈉為絡合劑,通過NaOH 調節pH,運用簡單的液相還原法制備出具有納米片組裝形貌的純相hcp-Co。Liu 等[43]以Ir 為成核劑、硬脂酸鈉為表面活性劑,運用溶劑熱多元醇法制備Co 納米棒,得到的產物為純相hcp-Co;通過提高Ir/Co 摩爾比來形成更多的Co 晶核,可縮短棒長度。Abel 等[44]在200~270 ℃和N2/H2氣氛下,用NaBH4還原溶解于四氫二甲基甲酰胺溶液中的CoCl2,得到的Co 納米顆粒均呈現hcp晶型,還可通過添加不同的表面活性劑來控制顆粒的大小。Viola 等[45]在175 ℃、NaOH 調節pH 的條件下,用1,2-丁二醇還原Co(CH3COO)2,得到hcp 和fcc 兩種晶型共存的球形Co 納米顆粒;而在反應前加入少量RuCl3時,則得到由純hcp-Co 納米顆粒構成的棒狀產物。這些方法豐富了hcp-Co 的制備手段,但還需將hcp-Co 納米粒子負載到載體上,以克服納米粒子易團聚的現象,才能應用于F-T 合成反應。
3 Hcp-Co 高活性的理論研究進展
3.1 B5 型位點
CO 解離的活化能對表面結構非常敏感,具有低活化能壘的結構處于階梯邊緣,也稱為B5 型位點,它通常由同一個平面中的四個原子和不同平面中的第五個原子形成;該位點本身不對應于任何特定類型的表面原子,而是由特定原子構成的空間。Santen 等[46-47]實驗研究發現,對于CO 解離,B5 位點表現的比表面Co 更加活躍,CO 以更高的速率解離,產生的C 原子快速加氫形成可移動到反應中心其他位置的CHx中間體來實現C—C鏈增長,從而防止焦炭形成。Ravi 等[48]使用嵌入原子勢法對具有不同粒徑的Co 納米粒子(2,4,6,10,15 nm)進行了經典的分子動力學模擬,發現在hcp-Co 和fcc-Co 納米顆粒中B5 型位點的分布有所差異,如圖2 所示。由圖2 可知,hcp-Co 和fcc-Co 上都存在B5-A 和B5-B 兩種位點,而B5-B’和B5-C 兩種位點僅存在于hcp-Co 上,它們對CO 吸附的空間阻礙較小、具有較強的CO吸附能力,這可能是hcp-Co 與fcc-Co 納米顆粒的催化活性差異的原因;hcp-Co 納米顆粒中較高的平臺原子與階梯/扭結原子的比率也可能是hcp-Co 納米顆粒性能更好的有利因素。Liu 等[7]基于DFT 的計算結果表明,hcp-Co 的高反應活性是由于它的晶體學對稱性,存在更多更活躍的B5 位點;hcp-Co 具有直接解離CO 的優勢,fcc-Co 需要H輔助解離CO,hcp-Co 比fcc-Co 更有利于CO 的解離活化。

圖2 不同粒徑下hcp-Co 和fcc-Co 納米顆粒中總B5 位點占表面原子數的比例(a)及10 nm 粒徑下hcp-Co 和fcc-Co 納米顆粒中各B5 位點類型占表面原子數的比例(b)[48]Fig.2 Proportion(a) of total B5 sites to the number of surface atoms in hcp-Co and fcc-Co nanoparticles with different particle sizes and proportion(b) of each B5 site in hcp-Co and fcc-Co nanoparticles with particle size of 10 nm[48].
3.2 hcp-Co 各晶面的鏈增長機理
在F-T 合成反應中,Co 基催化劑上的C—C鏈增長機理對Co 晶面敏感。Su 等[49]發現在較低的CO 覆蓋度下,鏈增長傾向于通過緊密堆積的hcp-Co(0001)晶面和階梯式Co 上的CO 插入機理進行,CH4是主要產物;碳化物機理在更開放的hcp-Co(10-11)晶面上是優選的,對C2烴的選擇性高于CH4。F-T 合成的結構靈敏度可歸因于吸附最不“飽和”中間體(如C 和CH)的結構靈敏度,最大程度地受益于結構演變;隨著CO 覆蓋度的增加,CO 插入機制變得更加有利,且hcp-Co(0001)晶面上的F-T 合成活性和C2烴選擇性都增加。Wen 等[50]通過DFT 計算研究了hcp-Co(0001)晶面上的C—C 鏈的引發、生長和終止機制。結果表明CHx(x=1~3)的形成比CH3OH更容易,CH 和CH2物種都主要與CHO 相互作用形成CHCHO 和CH2CHO,實現初始C—C 鏈形成,CHCHO 和CH2CHO 優先選擇連續加氫生成CH3CHO,然后發生C—O 鍵裂解得到CH3CH;隨后,CHO 插入CH3CH 可以實現進一步的鏈增長,即hcp-Co(0001)晶面上C—C 鏈增長是通過CHO插入實現的,C—C 鏈終止主要取決于金屬表面上的氫覆蓋度。Liu 等[51]將DFT 計算與微動力學模型用于探討hcp-Co(10-11)晶面上烴C—C鏈生長機理,通過碳化物機理的CH 和CH2自耦合實現了初始C—C 鏈形成,而不是通過CHO 插入機制;提出C—C 鏈增長機制是RCH2CH2與CH2偶聯形成R′CH2CH2(R′=RCH2);CHO 插入RCH2CH 形成RCH2CHCHO,然后加氫生成醇。微動力學模型顯示應考慮CH4形成對烴生成的影響,而醇對烴的選擇性的影響可忽略不計。結果證實hcp-Co(10-11)晶面顯示出更好的催化活性和對烴形成的選擇性。Hcp-Co(10-10)晶相鄰層間的距離或長或短,不同層間距終端的表面分別被定義為hcp-Co(10-10)-A 和hcp-Co(10-10)-B,其中hcp-Co(10-10)-A 的表面穩定性更好,且C 原子吸附強度弱于hcp-Co(10-11)晶面,因此hcp-Co(10-10)-A 晶面對催化F-T 合成反應更有優勢。
Zhang 等[52]對hcp-Co(10-10)-A 晶 面 進行了研究,證明了在hcp-Co(10-10)-A 晶面上CH2是最優選的CHx單體,C2烴主要通過碳化物機理形成;RCH2與CH2偶聯至R′CH2(R′RCH2),然后與CH2偶聯至R"CH2(R"R′CH2)實現C—C鏈生長循環以形成高級烴。
4 結語
由于hcp-Co 穩定存在的溫度低于fcc-Co,因此一些催化劑制備工作是通過添加有利于Co 還原的助劑,或對載體改性以抑制Co 與載體間的相互作用來降低還原溫度,從而促進 hcp-Co 的形成;運用RCR 活化步驟或控制氧化態Co 處于特定Co前體、再定向還原成亞穩態hcp-Co,也發展成為成熟的催化劑制備方法。通過實驗和理論計算研究,hcp-Co 基催化劑比fcc-Co 基催化劑有著更高的F-T 合成活性的原因可歸結為:Co 基催化劑應用于F-T 合成反應時,它的反應溫度低于250 ℃,有利于hcp-Co 的穩定存在,不易燒結失活;hcp-Co 有多種類型的B5 型位點和高活性晶面,使得催化劑對CO 的吸附和解離能力更強;hcp-Co 特定活性晶面的C—C 鏈增長能力優于fcc-Co。Co基催化劑的未來發展方向將是通過理論計算的支撐和實驗方法的改進,制備出暴露特定高活性晶面的高純度hcp-Co,以提高Co 基催化劑的F-T 合成反應性能。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
