運動神經元病合并額顳葉癡呆1例報告并文獻復習
2019-11-09 02:19:20劉江峰
中風與神經疾病雜志 2019年10期
關鍵詞:基因突變
劉江峰
運動神經元病(Motor neuron disease,MND)是一組以上和/或下運動神經元損害,主要表現為肌無力和萎縮、延髓麻痹和錐體束征的神經系統變性疾病;額顳葉癡呆(Frontotemporal dementia,FTD)是一組以明顯人格、行為改變和語言障礙為特征的癡呆綜合征。隨著人們對疾病的認識和研究的深入,不斷有運動神經元病合并額顳葉癡呆的個案報道,且發現兩種疾病可能存在相似的發病機制。為提高對本病的認識,現將我院近期收治的1例運動神經元病合并額顳葉癡呆病例報道如下。
1 臨床資料
患者,男性,62歲,因“進行性精神行為異常3 y,言語不清1 y余”于2019年4月22日入院。患者3 y前無明顯誘因出現精神行為異常,表現為重復講一些話、講他人壞話,打牌時經常多拿或少拿牌,并逐漸出現執行力、理解力、計算力、溝通能力、記憶力減退,常常答非所問,深夜無故外出,強迫性飲食,穿衣服時不能按順序穿,性格由原來暴躁變得溫和,于當地醫院就診,診斷為“老年性癡呆”,未予以特殊治療。1 y前患者無明顯誘因出現言語不清,伴雙上肢遠端肌肉萎縮,雙上肢乏力、持物不穩、飲水嗆咳。病程中無頭痛、頭暈及嘔吐、無肢體麻木、無隨地大小便等行為。既往體健,無高血壓及糖尿病病史;每天吸煙1包,嚼檳榔2包;嗜酒40余年,每天2~3 兩,已戒酒1 y。無家族遺傳疾病史。
查體:腋溫:36 ℃,呼吸:21次/min,脈搏:87次/min,血壓:121/95 mmHg。查體欠合作,神清,言語不清,慢性病容,注意力不集中,執行力、計算力、理解力、溝通能力均不同程度減退,記憶力損害較輕,視空間能力尚保留,頸軟,雙側瞳孔等大等圓,直徑約3 mm,對光反射靈敏,伸舌居中,咽反射正常,舌肌震顫、舌萎縮,雙上肢骨間肌、大小魚際肌、蚓狀肌萎縮(見圖1),上臂、前臂、肩胛帶肌群未見明顯萎縮,未見肌束顫動;雙上肢近端肌力5級、遠端肌力5-級,雙下肢肌力5級,四肢肌張力正常,雙側膝反射減弱,余深反射均可引出,霍夫曼征等病理征均(-);深淺感覺正常,共濟運動:指鼻試驗不配合,跟膝脛試驗不配合,閉目難立征(-)。輔助檢查:血常規、大便常規+OB、尿常規、心肌酶、肝腎功能、血糖血脂、電解質、凝血常規、輸血前4項、腦脊液常規、腦脊液生化、腦脊液細胞學、心電圖、胸片、腦電圖均正常。MMSE評分:9分。頭部MRI+MRA(見圖2):腦萎縮(以額顳葉為主),頭部MRA未見異常。肌電圖:雙側小指展肌、拇短展肌、第一骨間肌、指總伸肌、肱二頭肌、胸10椎旁肌、股內肌、脛前肌、腓腸肌、趾短伸肌均可見大量誘發電位、MUP明顯寬大,呈單時相或混合期,結合神經速度提示:呈廣泛性神經源性損害,支持運動神經元病診斷。
該患者病程3 y,雙上肢遠端肌萎縮、肌無力,構音障礙、舌肌萎縮、飲水嗆咳、強哭強笑,肌電圖提示廣泛性神經源性損害,根據運動神經元病El Escorial[1]診斷標準,可擬診為運動神經元病。此外,患者存在精神行為異常、認知功能減退,性格改變,飲食改變,執行力、理解力、計算力、溝通能力減退,記憶力損害較輕,視空間能力尚保留,MMSE評分低于正常,頭部MRI提示額顳葉萎縮,根據Neary等[2]制定的診斷標準,可臨床診斷為額顳葉癡呆,確診需行活檢或死后尸檢。故本例患者可診斷為運動神經元病-額顳葉癡呆綜合征。
2 討 論
運動神經元病(MND)是一組主要累及錐體束、腦干運動神經核、脊髓前角細胞的神經退行性疾病,通常在癥狀出現后3~5 y內死亡,約90%的MND病例呈散發性,10%的MND為家族性。近年來,越來越多人認為MND是一種可伴有認知障礙和行為改變的多系統疾病,約10%~15%的MND患者符合額顳葉癡呆的診斷標準[3],FTD癥狀可在運動癥狀之前、之后或同時出現。以下就MND-FTD的遺傳和病理機制、臨床特征、治療及預后進行討論。
2.1 發病機制 MND-FTD目前尚無確切發病機制,已有研究發現某些基因突變可引起MND,如銅/鋅超氧化物歧化酶(Cu/Zn superoxide dismutase 1,SOD1)基因突變、FUS基因突變、TARDBP基因突變等[4];而MAPT基因突變、GRN基因突變等[5]可引起家族性額顳葉癡呆,但這些突變基因都不能解釋MND-FTD。運動神經元病、額顳葉癡呆共病患者越來越多,這提示兩者可能存在共同的發病機制。DeJesus-Hernandez M和Renton AE等人發現C9orf72基因第一個內含子的六核苷酸GGGGCC擴增是家族性運動神經元病和額顳葉癡呆最主要的遺傳原因[6,7]。在家族性MND-FTD患者中,多達50%~70%的患者攜帶C9orf72基因重復擴增[8]。C9orf72基因重復擴增原因尚不明確,目前認為DNA高甲基化可能是C9orf72運動神經元病-額顳葉癡呆疾病的一個主要致病機制[9]。C9orf72基因重復擴增致病可能通過以下3種機制[10,11]:(1)雙向轉錄產生包含G4C2的RNA和G2C4的反義RNA,能夠隔離蛋白質,從而干擾其生理功能;(2)非AUG啟動子重復啟動翻譯產生二肽重復蛋白質(DPR),DPR的形成和累積可產生細胞毒性;(3)C9orf72基因外顯子序列轉錄減少可引起其單倍體功能不全。此外,有文獻報道,在少數MND-FTD患者或家族中發現了VCP、UBQLN2、OPTN和SQSTM1基因突變[12],這說明家族性MND-FTD存在著多種致病基因遺傳(即寡基因遺傳)。
神經元胞質異常蛋白的聚集是大多數神經退行性疾病的重要特征,有研究發現,約40%~50%的額顳葉癡呆、75%~90%的運動神經元病患者存在TDP-43病理包涵體[8],TDP-43異常蛋白質沉積是FTLD-TDP、MND的病理特征。Mackenzie IR等人根據TDP-43病理形態及分布的不同,將FTLD-TDP分為以下4種亞型[13,14]:A型:以新月形或橢圓形神經元胞質內含物(NCI)和多而短的營養不良性神經元突起(DN)為特征,主要位于上皮質,該亞型與GRN基因突變相關;B型:NCI數量中等,DN數量很少,分布于整個皮質層,MND-FTD及C9orf72基因突變與B型關聯最強;C型:NCI數量很少,DN數量多且長,主要分布于上皮質;D型:以神經元核內包涵體、短DN為主,NCI數量少,主要位于上皮質,該亞型與VCP基因突變臨床表型具有獨特相關性。除此之外,額顳葉癡呆變性(FTLD)還與tau蛋白、肉瘤融合蛋白(FUS)等有關[5]。運動神經元病的TDP-43蛋白在腦內的沉積模式與額顳葉癡呆不同,Brettschneider J[15]等人根據pTDP-43病理蛋白分布及解剖,提出MND病理分期:第一階段:pTDP-43蛋白主要累及運動皮質、腦干運動神經核及脊髓運動神經元等遠動系統;第二階段:累及額中回、腦干網狀核、小腦前核、紅核;第三階段:累及額葉前部、皮質感覺區及紋狀體;第四階段:累及顳葉內側及海馬。這表明MND是一種既選擇性影響運動系統、又影響其他神經元和膠質細胞的多系統神經退行性TDP-43蛋白病[14]。但是否所有的運動神經元病病理過程都以同樣方式進展,目前尚不清楚。
2.2 臨床特征 MND是一種進行性成人發病的神經退行性疾病,疾病類型主要包括肌萎縮側索硬化(ALS)、進行性延髓麻痹(PBP)、進行性肌萎縮(PMA)和原發性側索硬化(PLS),ALS是最常見的MND類型,PBP、PMA、PLS最終可進展為ALS。MND因疾病類型不同,而表現出不同的臨床癥狀組合,主要表現:構音障礙、吞咽困難、飲水嗆咳、流涎、肢體萎縮和無力、肌束顫動、呼吸困難等,以及病理征陽性、腱反射亢進、強哭強笑、肌張力增高等體征。FTD是一種具有明顯臨床表型的異質性疾病,其類型主要包括行為異常型額顳葉癡呆(bvFTD)、語義性癡呆(SD)、進行性非流利性失語(PNFA)。bvFTD主要表現在行為、個性、情感、執行控制力等方面的變化,這些變化包括抑制解除、冷漠、缺乏同情心、強迫/刻板行為、飲食變化等,bvFTD行為癥狀改變主要與額葉內側、額島葉皮質、扣帶回前部功能障礙有關[5];SD以單字理解障礙和命名性失語為主要特征,患者言語流利、語法正確,但不能理解單詞含義、找詞困難,SD主要與優勢半球顳葉受累有關[5]。PNFA主要表現為語言表達及交流能力下降,找詞困難,言語減少,理解力、記憶及視空間能力通常保留,PNFA與優勢半球額下回Broca區及島葉前部受累有關[5]。
MND的臨床癥狀可發生于FTD患者,同時MND在病程中也可表現語言或行為損害癥狀。Long Z[16]等發現大部分MND-FTD患者有語言障礙,且具有高度普遍及異質性等特點,其語言障礙包括失語癥、單字理解障礙、句子理解障礙、語法缺陷,MND-FTD語言障礙嚴重程度與FTD的語言表型相似。一項涉及279名散發性MND/FTD患者的研究中[17],約50%的MND患者存在認知障礙,10%~15%患者符合FTD標準,且主要表現為bvFTD。Cortes-Vicente E[18]等人在22例MND-bvFTD患者隨訪期間發現選擇性上肢遠端肌無力和萎縮伴不明顯下肢無力是主要的運動損害特征,嚴重的吞咽困難和吸入性肺炎是最常見的死亡原因,沒有癡呆的患者多數死于呼吸衰竭,約1/3患者一生中只表現為單純的下運動神經元(LMN)癥狀,從未出現上運動神經元(UMN)癥狀,但患者行基因檢測或死后接受神經病理檢查,卻支持MND診斷。
2.3 治療及預后 目前,暫無特效方法治療MND-FTD,雖然利魯唑在MND治療效果已被證實,但目前沒有研究直接檢測利魯唑在MND-FTD中的作用;FDA批準治療阿爾茨海默病的藥物對FTD也沒有獲益,且有研究表明乙酰膽堿酯酶抑制劑可能會加重FTD癥狀[19],美金剛在FTD的治療效果尚不明確。對于伴有躁動、易激惹、攻擊性行為的MND-FTD患者使用抗精神病藥物可能有用[8]。MND-FTD具有高致死性,預后差,有研究表明,MND-FTD患者疾病進展比沒有癡呆的MND進展更快[3],年齡大、延髓起病、存在認知功能損害、教育程度低的患者生存時間更短、預后更差[8,17]。
2.4 小結 綜上,雖然MND-FTD在遺傳和病理方面取得較大進展,但是否所有MND或FTD患者均有進展為MND-FTD的風險,或存在某些易感因素導致一些患者更易患MND-FTD,目前尚不明確。因MND-FTD的高致死率及致殘率,目前迫切需要針對該疾病基本機制的有效治療方法,針對C9orf72基因轉錄和翻譯等方面進行干擾可能是MND-FTD的治療靶點之一,但C9orf72基因及其他致病基因的發生和發展機制有待進一步闡明。目前有實驗對C9orf72基因轉錄及翻譯等方面進行研究,但尚未取得明顯進展。MND-FTD目前無統一的診斷標準,多數人采用同時符合MND和FTD各自標準來診斷這一疾病。未來,對MND-FTD致病機制及診斷標準進行深入研究將有利于治療和預后,為MND-FTD患者的有效治療帶來曙光。

圖1 患者雙側骨間肌、大小魚際肌、蚓狀肌萎縮
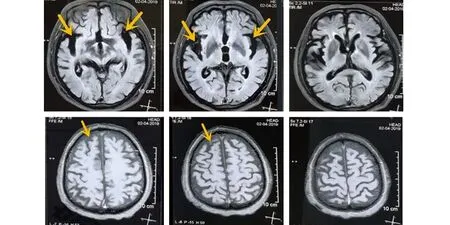
圖2 雙側額顳葉萎縮,以顳葉萎縮明顯
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
