X連鎖淋巴組織增生綜合征-2型1例報告并文獻復習
2020-04-11 03:43:20
精準醫學雜志 2020年1期
關鍵詞:基因突變
(青島大學附屬醫院,山東 青島 266003 1 血液兒科;2 藥學科)
X連鎖淋巴組織增生綜合征-2型(XLP-2)即X凋亡抑制因子缺乏癥,是一種罕見的X連鎖原發性免疫缺陷,主要表現為EBV驅動的噬血細胞綜合征(HLH),以發熱、脾腫大多見,NK細胞活性多正常。因病情發展迅速,若不及時診治預后較差。現將我院血液兒科診治的1例XLP-2患兒報告如下,并復習相關文獻,以提高臨床醫生對該病的認識。
1 臨床資料
患兒,男,3歲,因“反復發熱20余天”于2018年5月17日入院。患兒以弛張高熱起病,伴咳嗽,就診外院,查血常規示血紅蛋白106 g/L,余項均正常;谷草轉氨酶149.14 U/L,三酰甘油2.63 mmol/L,纖維蛋白原1.91 g/L,血清鐵蛋白10 600 μg/L;EBV DNA 1.31×109/L,EBV IgM陽性;腹部超聲檢查示肝脾大,雙肺CT檢查示雙肺高密度斑片影。每次給予患兒美洛西林鈉舒巴坦鈉75 mg/kg,每日2次,連用10 d,每天給予甲強龍1 mg/kg,連用5 d,一次性給予丙種球蛋白0.5 g/kg治療,患兒咳嗽好轉,因體溫不降轉入我院。既往史、個人史、家族史無特殊情況。
入院查體示體溫39 ℃,心率138 min-1,呼吸31 min-1,體質量15 kg。精神差,輕度貧血貌。全身無皮疹,頸部可觸及直徑2 cm×2 cm大小淋巴結。雙肺呼吸音粗,未聞及干濕性啰音。心臟查體未見異常。腹軟,肝臟肋下4 cm,脾臟肋下2.5 cm,質韌,無壓痛。輔助檢查:①血常規檢查血紅蛋白98 g/L,余項均正常;②三酰甘油3.01 mmol/L,纖維蛋白原1.25 g/L;③骨髓常規檢查:骨髓增生明顯活躍,未見吞噬細胞;④外周血NK細胞在刺激前后的細胞膜CD107a分子表達之差為2.43%,T細胞在刺激前后的細胞膜CD107a分子表達之比為1.8;CD3-CD56+NK細胞占淋巴細胞的比例為3.69%,NK細胞顆粒酶B陽性率為70.34%,NK細胞穿孔素陽性率為94.68%;⑤血清鐵蛋白740.89 μg/L;⑥可溶性CD25(sCD25)2 016.28 ng/L;⑦EBV IgM陽性;⑧外周血NK細胞和T細胞XIAP表達率與同型對照組之差分別為58%、54%;⑧細胞因子:IL-1β 12.3 ng/L,IL-6 20.6 ng/L,IL-18 8.2 ng/L;⑨家族性噬血細胞綜合征相關二代基因測序結果顯示,患兒XIAP基因c.1141C>T(p.R381X)(圖1A),患兒父親無變異(圖1B),患兒母親檢測到該處的雜合變異,未檢測到編碼區的堿基異常改變(圖1C);⑩胸部CT檢查未見異常,腹部CT檢查示肝脾腫大。
診斷和治療:本例患兒存在XIAP基因突變,發熱超過5 d,脾腫大,高三酰甘油血癥,低纖維蛋白原血癥,血清鐵蛋白升高,診斷為XLP-2。治療經過:按照噬血細胞綜合征-2004(HLH-2004)方案給予地塞米松治療3 d后患兒體溫降至正常,治療2周評價療效,EBV DNA陰性,三酰甘油、纖維蛋白原正常,脾臟回縮,肝臟無回縮,行B超引導下肝臟穿刺活檢病理結果示肝小葉結構尚清,肝細胞重度水腫,匯管區少量炎細胞浸潤,未見明確噬紅細胞現象,EBV編碼的小RNA陰性。治療4周血清鐵蛋白降至正常,肝臟回縮至正常,遂出院。治療8周激素減停。停藥16周后復查NK細胞脫顆粒功能異常,NK細胞顆粒酶B陽性率低,建議進行造血干細胞移植(HSCT),家屬拒絕,定期門診隨訪。目前患兒XLP-2未復發,查血常規、EBV-DNA正常。
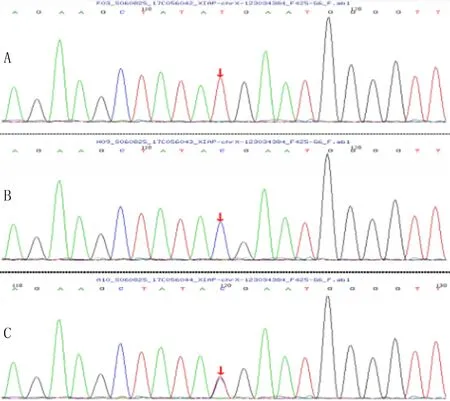
A、B、C分別為患兒、患兒父親、患兒母親
2 討 論
XLP是最經典的EBV驅動的HLH,包括有XLP-1和XLP-2兩種類型,分別對應著SH2D1A及XIAP/BIRC4兩種基因突變[1-2]。XLP-2常表現為EBV驅動的HLH(76%)、克羅恩病(26%)以及低丙種球蛋白血癥(33%)等合并癥[3]。XLP-2中HLH嚴重程度較XLP-1低,極少合并神經系統癥狀及淋巴瘤[4]。一些XLP-2患者還表現為自身免疫性疾病,如關節炎、結節性紅斑和腎炎等[5]。本例患兒此次發病尚無克羅恩病、低丙種球蛋白血癥等合并癥,需警惕再次復發合并上述疾病的可能。
XLP-2是一種罕見的X連鎖原發性免疫缺陷疾病[6-7]。該突變由RIGAUD等[8]在2006年首次被發現,其發病率為(1~2)/1 000 000,多數為男性患者。XIAP/BIRC4基因位于X染色體長臂25區(Xq25),與SH2D1A基因相鄰,包含6個外顯子,編碼表達XIAP蛋白質。到目前為止,在XIAP患者中已經發現超過70種包括插入、缺失等造成的無義或錯義基因突變,這些突變沿外顯子分布,導致蛋白質表達的部分或完全喪失[9]。本例患兒的XIAP基因有1個半合子突變,且母親存在該位點的雜合突變,符合XLP-2的遺傳特點。已有文獻報道,該突變可引起XIAP蛋白UBA結構域改變,使肽鏈合成過早終止,蛋白表達喪失[10]。患兒NK細胞和T細胞XIAP蛋白表達降低,為XIAP基因缺陷引起的功能性蛋白表達降低。XIAP蛋白質是一種相對分子質量為56 000的凋亡抑制分子(IAPs),由含有三個桿狀病毒IAP重復結構域[11]、進化上相對保守的UBA結構域和RING結構域組成,與半胱天冬酶-3、-7和-9型結合,直接抑制其蛋白水解活性從而抑制細胞凋亡[12]。在正常個體中,淋巴細胞、NK細胞等均可見XIAP表達[13]。XIAP蛋白一方面通過抑制活化T細胞中的半胱天冬酶,減少CD8+T細胞的凋亡[14];另一方面,XIAP參與針對細菌和真菌感染的模式識別受體(NOD1/2和Dectin-1)的免疫調節[15]。當NOD1/2和Dectin-1在感染期間被其配體激活時會引起炎癥反應清除病原體[16-17]。XIAP缺乏可導致患者體內淋巴細胞過度活化[18],并使Toll樣受體觸發半胱天冬酶-8型介導的細胞壞死增加、IL-1β和IL-18分泌增多,繼而表現出HLH的癥狀[19-20]。
基因測序是確診XLP-2的金標準[8,15],NK細胞的活性檢測是目前常用的免疫學參數。不同于XLP-1,XLP-2患者NK細胞的活性是正常的[21-22]。XLP-1中的NK細胞不能殺死EBV感染的B細胞,這種NK細胞毒性缺陷涉及兩個淋巴細胞活化信號分子相關受體,即2B4和SLAMF6。這兩種受體可觸發和激活NK細胞和CD8+T淋巴細胞對EBV感染B細胞的細胞毒性[23]。已知B細胞淋巴瘤和EBV感染的B細胞中2B4的配體CD48表達會增加。在XLP-1中,2B4和CD48之間的相互作用致EBV感染的靶細胞裂解受到抑制。這也解釋了XLP-1患者易合并淋巴瘤,而XLP-2患者尚未發現該易感性原因[5]。不同于已報道的XLP-2患者,該患者NK細胞的數量及活性降低,于疾病轉歸后復查無改善,與XLP-1表型相似。這種相似性提高了XLP-1中缺失的信號轉導淋巴細胞激活分子相關蛋白(SAP蛋白)和XIAP蛋白之間功能表達的分子聯系;或兩者分別涉及不同的獨立通路,但均可在EBV感染的控制中發揮作用,導致共同的表型。
XLP-2的治療分為兩個方面,一方面是誘導緩解以控制炎癥反應進展,另一方面是病因治療糾正免疫缺陷以防止復發[2]。目前廣泛應用的誘導治療方案為HLH-2004,HSCT是XLP-2唯一治愈性治療方法[15]。然而,在一項對XLP-1和XLP-2患者的研究中發現,有或沒有HSCT的患者存活率均較低[24]。但XLP-2患者HLH易反復,如不進行HSCT,必須持續密切監測患者的疾病進展[25]。有文獻已報道1例XIAP基因突變者單用激素治療療效良好[26]。本例患者單用激素治療敏感,但患兒已發現XIAP基因突變,符合HSCT指征,但家屬拒絕行HSCT。患兒現隨訪時間內病情尚穩定,多次復查EBV-DNA陰性,需繼續動態檢測。
綜上所述,XLP-2因病情發展迅速,若不及時診治預后較差。采用針對性實驗室檢查和基因檢測有助于進一步明確XLP-2的臨床和免疫表型,從而指導個體化治療方案的實施。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
