基于實時熒光定量PCR檢測的沙門氏菌標準物質驗證
2020-05-11 02:00:28王菲張玲楊佳怡葉子弘俞曉平
食品與發酵工業 2020年8期
王菲,張玲,楊佳怡,葉子弘,俞曉平*
1(中國計量大學,浙江省生物計量及檢驗檢疫技術重點實驗室,浙江 杭州,310018)2(中國計量科學研究院,北京, 100013)
沙門氏菌(Salmonella)是一類革蘭氏陰性、具有鞭毛的兼性需氧菌[1],是細菌性食源性疾病的主要致病菌之一,易引起食物中毒以及腸胃炎等疾病[2-3]。所以針對食品原料、產品等進行沙門氏菌檢測是我國食品監管部門及食品相關企業的必檢項目[4-5]。傳統的沙門氏菌分離、檢測和鑒定方法主要有常規培養法、免疫學方法等[6],然而,這些方法操作較復雜、檢測周期較長,且需要消耗大量培養基和生化試劑,對檢測人員的技術水平也有較高的要求[7-8]。因此,隨著分子生物學技術的發展,越來越多的實驗室選擇實時熒光定量PCR技術進行沙門氏菌檢測[9-10]。實時熒光定量PCR技術具有操作簡單、耗時短、效率高等優點[11],但該技術能否準確檢測沙門氏菌還有待驗證。
本研究選擇沙門氏菌DNA標準物質候選物,使用實時熒光定量PCR技術來驗證標準物質的均勻性和穩定性。為確保數據的準確性,還設計不同PCR儀精度分析、不同酶體系比較以及添加小牛胸腺載體DNA作為保護劑分析等。本文對沙門氏菌的穩定性研究可為各實驗室進行比對實驗以及快速檢測方法研究提供基礎參考。
1 材料與方法
1.1 試劑與儀器
沙門氏菌菌株,中國工業微生物菌種保藏中心,凍存于-80℃冰箱。Fast TaqMan Mixture,北京康為世紀生物科技有限公司;SuperRealPreMix(Probe),天根生化科技(北京)有限公司;Premix Ex TaqTM(Probe qPCR),寶日醫生物技術(北京)有限公司(takara中國);TaqMan Gene Expression Master Mix,美國應用生物系統公司;小牛胸腺DNA(calf-thymus DNA,CT-DNA),上海澤葉生物科技有限公司;沙門氏菌DNA標準物質,中國計量科學研究院研制;引物和探針由英濰捷基(上海)貿易有限公司合成(表1)。
Q-POD純水儀,儀默克化工技術(上海)有限公司;XS205電子秤,梅特勒-托利多國際貿易(上海)有限公司;生物安全柜,美國賽博飛公司;超低溫保存箱,海爾生物醫療股份有限公司;3K15臺式高速冷凍離心,德國Sigma公司;AB quant Studio 12K Flex、Roche羅氏LightCycler?480Ⅱ實時熒光定量PCR儀,上海普迪生物技術有限公司。
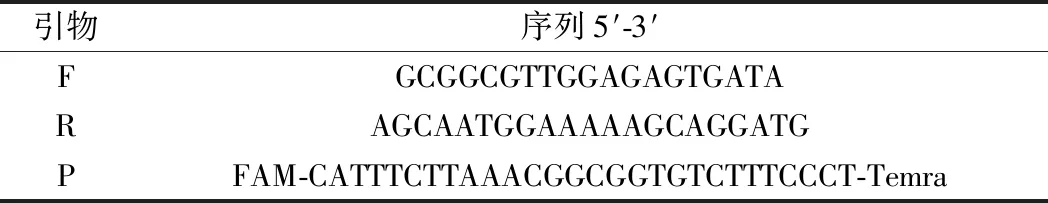
表1 PCR所用引物和探針Table 1 Primers and probes for PCR
1.2 實驗方法
1.2.1 沙門氏菌DNA模板制備
沙門氏菌DNA作為定量標準物質候選物,使用滅菌去離子水對沙門氏菌DNA進行10倍系列稀釋備用[12],最終得到的濃度分別為2.27×105copy/μL,2.27×104copy/μL,2.27×103copy/μL,227 copy/μL,22.7 copy/μL,2.27 copy/μL。
使用TE緩沖液(10 mmol/L Tris+0.1 mmol/L EDTA,pH 7.4)對小牛胸腺DNA溶液進行稀釋,得到終濃度為20 μg/mL。并使用稀釋過后的小牛胸腺DNA溶液對沙門氏菌DNA進行10倍系列稀釋,稀釋得到的最終濃度分別為2.27×105copy/μL,2.27×104copy/μL,2.27×103copy/μL,227 copy/μL,22.7 copy/μL,2.27 copy/μL。
1.2.2 PCR檢測體系的建立
PCR反應采用25 μL體系,2×PCR Mix 12.5 μL,引物F(10 μmol/L)1 μL,引物R(10 μmol/L)1 μL,探針P(10 μmol/L)1μL,DNA模板5 μL,ddH2O 4.5 μL。PCR反應程序為95 ℃預熱3 min;94 ℃ 5 s、60 ℃ 40 s,共40個循環,4 ℃保存反應產物。
1.2.3 實時熒光PCR檢測方法的特異性驗證
PCR引物探針的設計參考了腸道沙門氏菌腸毒素傷寒桿菌亞種,NCBI的Genebank序列號為NC_003198.1。特異性實驗采用的菌株如表2所示,所有菌株皆為本實驗室提供。將所有的菌株進行平板劃線培養,挑選單菌落加入50 μL的滅菌去離子水中,煮沸裂解后分別離心收集上清液作為模板,進行PCR擴增。滅菌去離子水作為空白對照,本研究使用的沙門氏菌DNA標準物質作為陽性對照,驗證沙門氏菌檢測時本研究建立的PCR檢測方法的特異性。
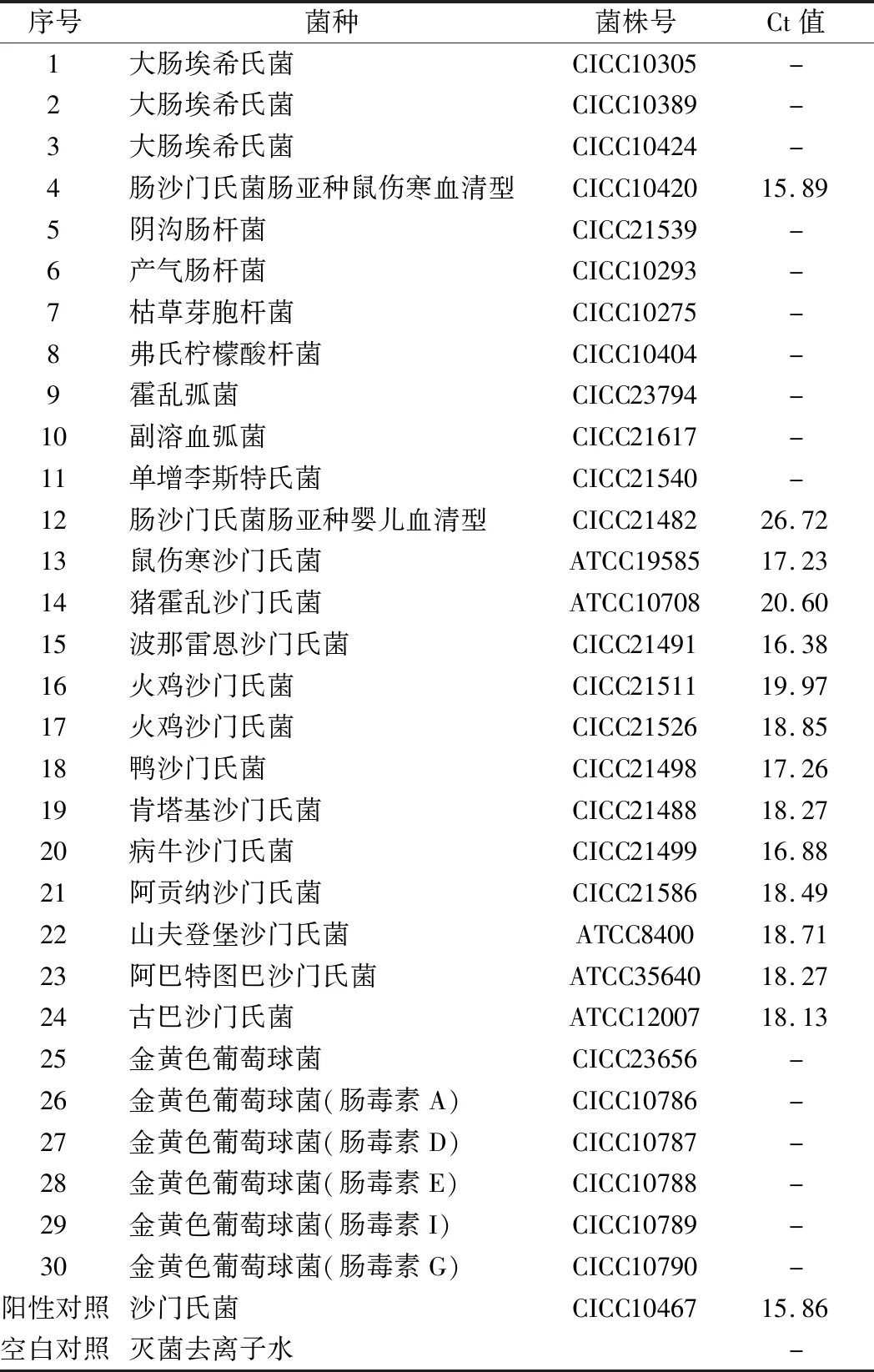
表2 沙門氏菌DNA的特異性檢測Table 2 Specific detection of Salmonella DNA
注:-代表無
1.2.4 不同PCR儀對沙門氏菌DNA熒光定量PCR的影響
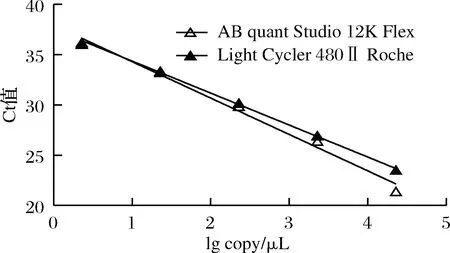
使用AB quant Studio 12K Flex和Light Cycler 480Ⅱ Roche實時熒光定量PCR儀分別對沙門氏菌DNA進行實時熒光定量PCR,以滅菌去離子水10倍系列稀釋的沙門氏菌DNA為模板,經實時熒光定量PCR后的Ct值,從斜率,線性相關數,標準偏差以及擴增效率方面來分析不同PCR儀對沙門氏菌DNA擴增的影響[13]。其中擴增效率的計算如公式(1)所示:
(1)
式中:E表示擴增效率,slope表示斜率。
1.2.5 TaqDNA聚合酶對沙門氏菌DNA熒光定量PCR的影響
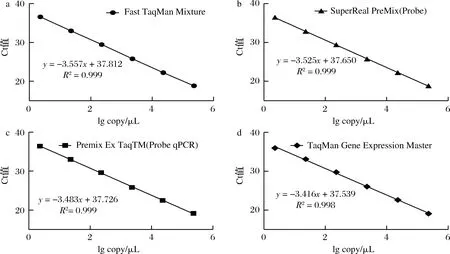
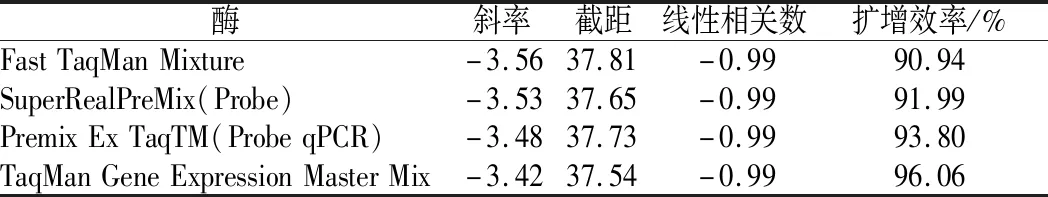
分別采用市面上常用的TaqDNA聚合酶Fast TaqMan Mixture、SuperRealPreMix(Probe)、Premix Ex TaqTM(Probe qPCR)、TaqMan Gene Expression Master Mix對沙門氏菌DNA進行實時熒光定量PCR,以滅菌去離子水10倍系列稀釋的沙門氏菌DNA為模板,經實時熒光定量PCR后的Ct值,從斜率、截距、線性相關數以及擴增效率方面來分析不同的TaqDNA聚合酶對沙門氏菌DNA擴增的影響。
1.2.6 添加保護劑對沙門氏菌DNA熒光定量PCR的影響
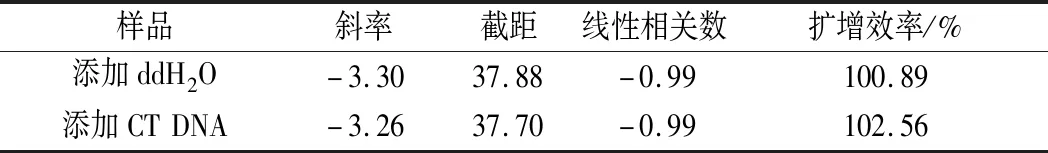
以10倍系列稀釋的沙門氏菌DNA為模板進行定量PCR(每組實驗重復3次),同時以去離子水作為空白對照,根據DNA濃度的對數和Ct值建立沙門氏菌的熒光定量PCR法標準曲線,從斜率、截距、線性相關數以及擴增效率方面來分析載體DNA是否會影響沙門氏菌DNA熒光定量PCR檢測。
1.2.7 沙門氏菌DNA均勻性測試
選取7組沙門氏菌DNA作為平行樣品,經實時熒光定量PCR儀測定Ct值,每組實驗重復3次,同時以滅菌去離子純水作為陰性對照,用Excel整理數據,計算由滅菌去離子水存放的低濃度沙門氏菌DNA的均勻性。其中均勻性分析使用方差分析法,如公式(2)所示:
(2)

1.2.8 短期穩定性測試
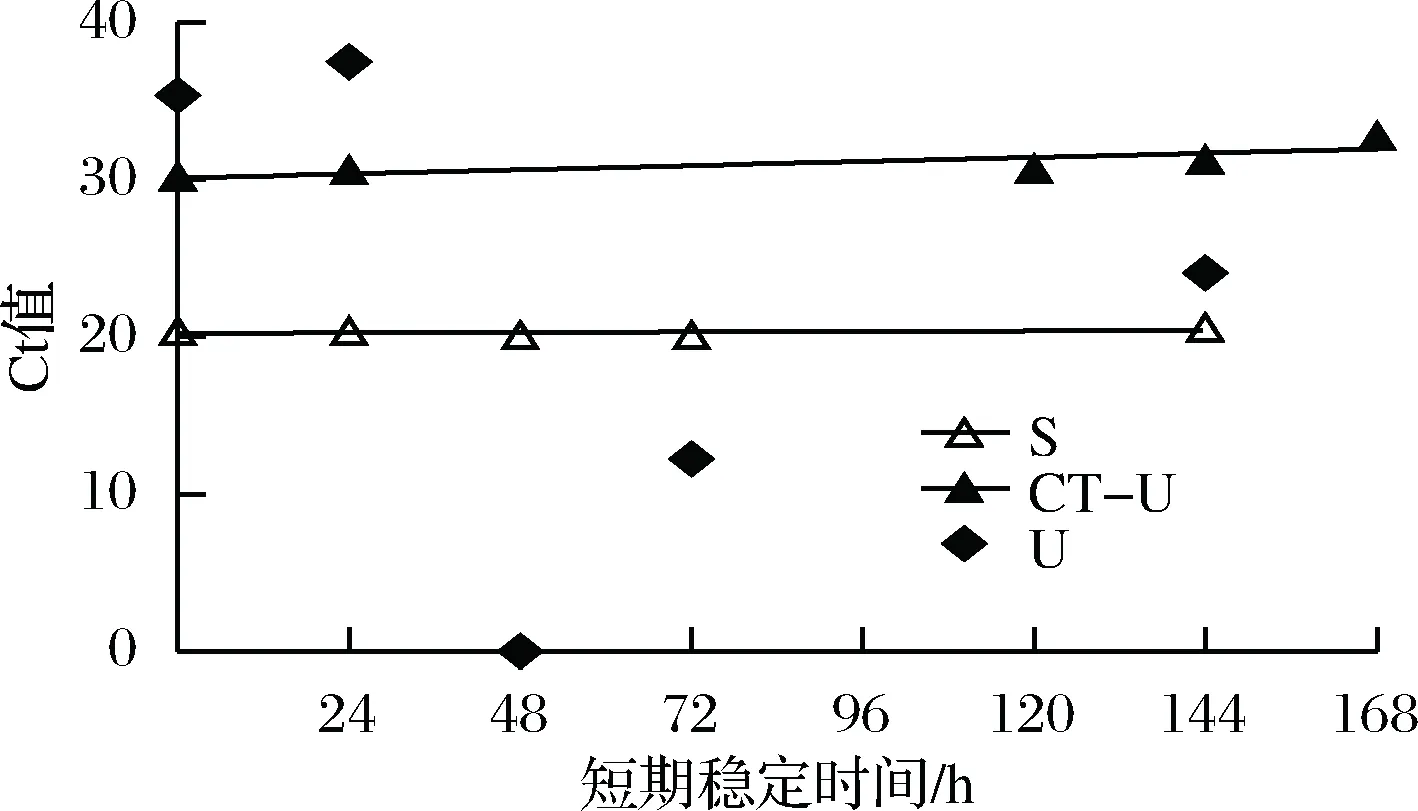
將沙門氏菌DNA標準物質候選物儲存于溫度為(40±2) ℃的烘箱中,設計存儲時間(0、24、48、72、144 h或0、24、120、144、168 h)[14],每個時間點包括3組平行樣品。使用實時熒光定量PCR儀測定Ct值(每組實驗重復3次),同時以去離子純水作為陰性對照,用Excel整理數據計算平均值,繪圖分析未稀釋的沙門氏菌DNA以及保護劑中存放的低濃度沙門氏菌DNA檢測樣的短期穩定時間。
其中重復性計算如公式(3)、公式(4)所示:
(3)
(4)
復現性計算如公式(5)~公式(7)所示:
(5)
(6)
(7)
公式(3)、公式(4)表示的是置信概率為95%時實時熒光定量PCR檢測方法的重復性,公式(5)~公式(7)表示的是檢測方法的復現性,其中m為平行樣品的數量,n為平行測定次數。
穩定性計算如公式(8)、公式(9)所示:
(8)
(9)
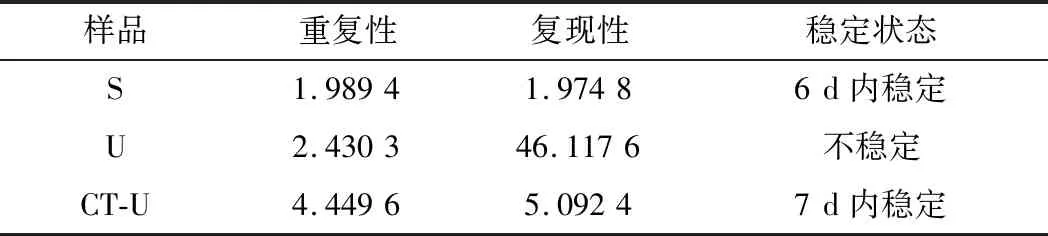
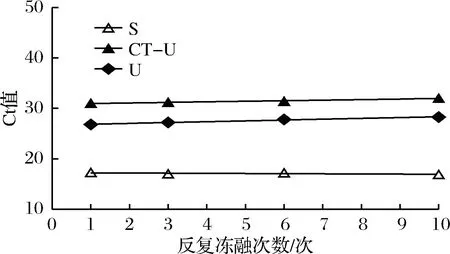
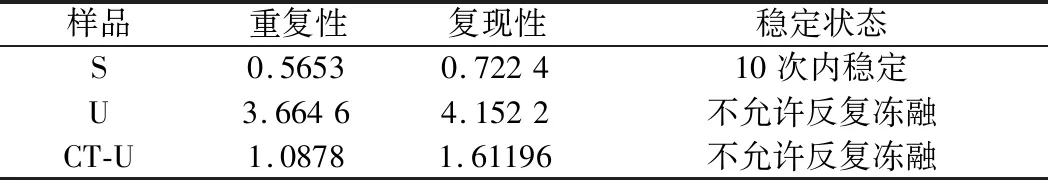
公式(8)表示的是直線的標準偏差,公式(9)表示的是斜率k的不確定度,自由度為n-2,95%置信水平的t分布因子查表可知,若|k| 1.2.9 反復凍融穩定性測試 將沙門氏菌DNA標準物質候選物放入冰箱(溫度-80 ℃,時間5 min),常溫下解凍(時間10 min),計劃操作次數(1、3、6、10次),每次操作取3個平行樣品放入4 ℃冰箱備用。使用實時熒光定量PCR儀測定Ct值(每個樣品重復實驗3次),同時以去離子純水作為陰性對照,用Excel整理數據計算平均值,繪圖分析未稀釋的沙門氏菌DNA以及保護劑中存放的低濃度沙門氏菌DNA檢測樣能夠經受溫度變化的次數。 1.2.10 數據分析 基本數據采用Excel 2013進行分析(計算平均值、斜率、截距、擴增效率、重復性、復現性),采用GraphPad Prism 5.0對不同研究條件的沙門氏菌檢測參數進行分析(方差分析法、直線擬合法、t檢驗統計檢驗法)[16]。 2.1.1 沙門氏菌熒光定量PCR檢測方法特異性分析 引物/探針組合對實驗所檢測各物種的特異性實驗結果顯示(表2),當使用引物和探針組合對31種DNA樣本進行擴增時,只有在沙門氏菌的DNA樣本出現擴增曲線,可以獲取Ct值,而在其他細菌種類中沒有出現擴增曲線。因此建立的沙門氏菌實時熒光PCR檢測方法具有高度的特異性。 2.1.2 不同PCR儀熒光定量PCR分析 根據實時熒光定量PCR技術檢測結果,對相關數據進行分析(表3),得到AB quant Studio 12K Flex和Light Cycler 480Ⅱ Roche兩臺PCR儀的線性分布圖(圖1)。從斜率和線性相關系數上分析,兩臺儀器的檢測結果相近,無明顯區別,均可用于沙門氏菌熒光定量PCR檢驗。 2.1.3 TaqDNA聚合酶熒光定量PCR分析 使用市面上常見的4種酶,對沙門氏菌DNA建立不同的酶體系進行Ct值分析(圖2),評估不同酶體系作用于沙門氏菌DNA的熒光擴增效率[公式(1)],基本輸出是一個線性關系,表示不同濃度梯度的沙門氏菌DNA的Ct值,相關統計量如表4所示。不同的酶相關系數約為0.99和擴增效率(在90%~100%)都處在實驗接受范圍內,表明這4種酶都適用于檢測沙門氏菌DNA,可根據自己的實驗條件,選擇和調整適當的酶體系。 圖1 不同PCR儀比較線性分布圖Fig.1 Comparison of linear distribution of different PCR instruments 表3 不同PCR儀實驗結果分析Table 3 Analysis of the experimental results of different PCR instruments a-Fast TagMan Mixture;b-SuperReal PreMix;c-Premix Ex TaqTM;d-TaqMan Gence Expressin Master圖2 不同酶比較實驗Fig.2 Comparison of different enzymes 表4 酶比較實驗參數分析Table 4 Analysis of experimental parameters of enzyme comparison 2.1.4 保護劑熒光定量PCR分析 通過添加保護劑與未添加保護劑的沙門氏菌Ct值的比較(圖3),用以驗證添加的保護劑小牛胸腺載體DNA是否影響沙門氏菌DNA的熒光擴增,相關統計量參數如表5所示。由表5可知,添加保護劑后的沙門氏菌DNA與為添加保護劑的沙門氏菌DNA熒光擴增后有相近的斜率,截距,線性相關數以及擴增效率[公式(1)],可知小牛胸腺載體DNA不會影響沙門氏菌DNA的PCR檢測。 a-添加ddH2O;b-添加CT DNA圖3 驗證實驗標準曲線圖Fig.3 Standard curve of validation experiment 表5 驗證實驗參數分析Table 5 Analysis of validation experimental parameters 由于沙門氏菌標準物質候選物包裝單位約為100個,因此,隨機抽取7個包裝單位作為均勻性檢測的樣品,每個樣品在不同的部位分別取3個試樣進行實時熒光定量PCR檢測。沙門氏菌均勻性檢測結果以及相關統計量如表6所示。 表6 沙門氏菌均勻性檢測結果Table 6 Detection results of homogeneity of Salmonella 均勻性檢驗使用的是方差分析法,檢測標準物質在規定細分范圍內其特性是否保持不變。根據表5和公式(2)可以計算出F=2.99,由F表可以查詢出F0.05(6,14)=3.96,可見F 標準物質的短期穩定性,主要關注的是標準物質運輸過程中的穩定性。一般情況下,運輸過程中的高溫環境對標準物質有較大的影響。在不同的存儲時間中,沙門氏菌Ct值的會產生不同程度的變化(圖4)。根據直線擬合法的計算分析[公式(3)~公式(9)],計算結果如表7所示,初始濃度的沙門氏菌DNA和保護劑中存放的低濃度沙門氏菌DNA量值是比較穩定的,但是由滅菌去離子水中存放的低濃度沙門氏菌DNA量值不穩定。 圖4 短期穩定性測試Fig.4 Short-term stability test 表7 短期穩定性結果分析Table 7 Stability analysis of Short-term 注:S,未稀釋過的沙門氏菌DNA;U,由滅菌去離子水存放的低濃度沙門氏菌DNA檢測樣;CT-U,由保護劑存放的低濃度沙門氏菌DNA檢測樣(下同) 反復凍融穩定性,主要關注的是標準物質在進行實驗分析過程中的穩定性[17]。進行反復凍融實驗時,沙門氏菌Ct值的變化情況可以呈線性分布(圖5)。但是根據直線擬合法的計算分析(表8),初始濃度沙門氏菌DNA經過數次反復凍融仍然穩定,可以進行多次重復實驗。但是低濃度的沙門氏菌DNA不允許反復凍融,為保證數據的可靠性,需對獲取的樣品進行一次性操作。 圖5 反復凍融穩定性測試Fig.5 Testing of the stability of repeated freezing and thawing 表8 反復凍融穩定性分析Table 8 Stability analysis of repeated freezing and thawing 本研究使用沙門氏菌DNA標準物質對實時熒光定量PCR檢測方法進行規范驗證[18-20],并選擇2臺不同的PCR儀[21]對檢測方法進行比較驗證。為優化PCR體系,選擇4種不同的酶[22]進行比較。在初步的實驗中,直接使用沙門氏菌DNA進行實驗表明沙門氏菌DNA并不穩定,所以為了降低沙門氏菌DNA的降解率,選擇使用小牛胸腺載體DNA[23]作為保護劑。 實驗顯示,在95%置信水平時,沙門氏菌標準物質樣品均勻。沙門氏菌標準物質的穩定性研究[24]主要分為兩個部分,一個是模擬短期高溫穩定性,另一個是反復凍融穩定性,分別對初始濃度沙門氏菌DNA、ddH2O保存的低濃度沙門氏菌DNA檢測樣以及小牛胸腺DNA緩沖液保存的低濃度沙門氏菌DNA檢測樣進行實驗。研究表明初始濃度沙門氏菌DNA檢測樣6 d內量值穩定,而且可以進行反復凍融10次,針對一個樣品進行多次實驗操作,不會影響實驗數據的準確性;由小牛胸腺DNA溶液保存的低濃度沙門氏菌DNA檢測樣7 d內量值穩定,但是進行反復凍融實驗時,沙門氏菌DNA結構遭到破壞未能測到有效數據。總的來說,這兩種濃度的沙門氏菌短期穩定時間長,作為標準物質在運輸過程中高溫影響小[25],但實驗時避免反復凍融。但是由ddH2O保存的低濃度沙門氏菌DNA檢測樣不管是短期穩定實驗還是反復凍融穩定實驗都表現出不穩定,建議實驗時對沙門氏菌DNA添加保護劑,降低沙門氏菌DNA的消耗。 在未來研究中將進行長期穩定性實驗并分析影響沙門氏菌降解的影響因素,為沙門氏菌穩定性研究提供參考資料。 實時熒光定量PCR方法[26-28]在病原微生物快速檢測應用中準確性高,且沙門氏菌DNA穩定性好,可用來檢驗各實驗室開展微生物核酸PCR方法的測量能力,保障微生物核酸檢測技術的溯源性和準確性,從而提高我國在病原微生物核酸快速檢測的水平。2 結果與分析
2.1 方法驗證結果分析

2.2 均勻性檢測分析

2.3 短期穩定性檢測分析
2.4 反復凍融穩定性檢測分析
3 結論
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58民用飛機設計與研究(2020年4期)2021-01-21 09:15:02小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50電子制作(2018年18期)2018-11-14 01:48:24山東工業技術(2016年15期)2016-12-01 05:31:22發明與創新(2016年38期)2016-08-22 03:02:52太空探索(2016年5期)2016-07-12 15:17:55
