木犀草素納米混懸劑的制備與質量評價
2020-06-23 06:32:26何佶彥
天津中醫藥 2020年6期
謝 慧,何佶彥
(湖北六七二中西醫結合骨科醫院藥學部,武漢 430070)
木犀草素(Luteolin)最早是從木犀草科草本植物木犀草(Resedaodorata L.)中分離出來的一種黃酮類化合物,目前已發現存在于菊科、唇形科、天南星科、十字花科、馬鞭草科等植物中。最初研究發現木犀草素具有以抗炎,抗氧化,保護神經系統,改善記憶力等藥理作用[1],隨著研究的深入,發現對木犀草素具有抑制腫瘤細胞增殖,誘導腫瘤細胞凋亡等藥理活性[2]。然而,木犀草素在水中溶解度極低(約為61 μg/mL),影響藥物體內吸收,口服生物利用度較低[3-4],限制了臨床應用。為了提高木犀草素的溶解度及藥效,研究人員將其制備成聚合物納米粒[5]、白蛋白納米粒[6]、脂質體[7]、固體分散體[8]等新型給藥系統。納米混懸劑是近年來開發的一種納米給藥系統,由于其粒徑較小,存在巨大的比表面積和界面能,能夠顯著增加難溶性藥物的溶解度及口服生物利用度[9-10]。因此,本研究將木犀草素制備成納米混懸劑,通過單因素實驗優化了制備工藝參數并確定了處方中穩定劑和表面活性劑的種類,并最終采用Box-Behnken實驗設計優化得到木犀草素納米混懸劑的最優處方,為木犀草素的動物體內藥動學研究奠定實驗基礎。
1 儀器與材料
ESW-1.0小型濕法研磨機(上海易勒機電設備有限公司);氧化鋯(ZrO2)珠研磨介質(直徑為0.5 mm);Zetasizer Nano ZS90 納米粒徑/電位分析儀(英國馬爾文公司);JSM-7800F掃描電鏡(日本電子公司);iChrom 5100系列高效液相色譜儀(大連依利特分析儀器有限公司);RCY-1400T智能溶出試驗儀(天津市瑞斯德科技有限公司)。
木犀草素原料藥(西安唯奧生物科技有限公司,純度:98.5%,批號:20180923);木犀草素對照品(成都曼思特生物科技有限公司,純度:99.5%,批號:MUST-18061407);羥丙基纖維素(HPC SL,日本信越化學);聚維酮(PVP K30,巴斯夫輔料公司);羥丙甲纖維素(HPMC E5,陶氏化學);泊洛沙姆(Poloxamer 188,巴斯夫輔料公司);維生素E聚乙二醇1000琥珀酸酯(TPGs,巴斯夫輔料公司);吐溫80(Tween-80,南京威爾藥業股份有限公司)。
2 方法與結果
2.1 木犀草素納米混懸劑的制備 通過介質碾磨法[11]制備木犀草素納米混懸劑。稱取一定量的穩定劑(HPC SL、PVP K30或 HPMC E5)加入到純化水中溶解,再加入表面活性劑(Poloxamer 188、Tween-80或TPGs)溶解,備用。稱取一定量木犀草素加入到上述溶液中,分散,并加入到介質碾磨機中,加入一定量ZrO2,以一定的速度進行碾磨一定時間,取樣測定粒徑分布和Zeta電位,制備木犀草素納米混懸劑。
2.2 粒徑分布及Zeta電位測定 取木犀草素納米混懸劑加入少量蒸餾水稀釋,用Zetasizer Nano ZS90納米粒徑/電位分析儀測定木犀草素納米混懸劑的粒徑分布和Zeta電位,每份樣品重復測定3次。
2.3 處方工藝篩選
2.3.1 穩定劑種類篩選 文獻報道[12]納米混懸劑處方中加入穩定劑及表面活性劑可以提高體系的穩定性。本研究固定藥物濃度為30 mg/mL,碾磨介質與混懸劑體積之比為1∶1,碾磨速度為2 500 r/min,碾磨時間為4 h,每個處方中分別加入濃度為2%的HPC SL、PVP K30和HPMC E5作為穩定劑制備木犀草素納米混懸劑,測定粒徑分布及Zeta電位,篩選穩定劑。研究結果表明,不同種類的穩定劑會對木犀草素納米混懸劑的粒徑及Zeta電位大小產生一定的影響,使用HPC SL作為穩定劑制備的納米混懸劑粒徑分布最小,Zeta電位較低,因此本研究使用HPC SL作為穩定劑制備木犀草素納米混懸劑。
2.3.2 穩定劑濃度篩選 固定藥物濃度為30mg/mL,碾磨介質與混懸劑體積之比為1∶1,碾磨速度為2 500 r/min,碾磨時間為4 h,在處方中分別加入濃度為 0.5、0.75、1、2、5%的 HPC SL 制備木犀草素納米混懸劑,測定粒徑分布及Zeta電位,篩選穩定劑濃度。研究驗結果表明,穩定劑濃度對木犀草素納米混懸劑的粒徑及Zeta電位產生一定的影響,隨著穩定劑濃度的增大,制備的納米混懸劑粒徑增大,Zeta電位隨著穩定劑的濃度增加而增大,因此穩定劑的濃度需要進一步優化。
2.3.3 表面活性劑種類篩選 文獻報道[13]納米混懸劑中的表面活性劑種類會對其粒徑及穩定性產生一定的影響。固定藥物濃度為30 mg/mL,碾磨介質與混懸劑體積之比為1∶1,碾磨速度為2 500 r/min,碾磨時間為4 h,HPC SL濃度為2%,處方中分別加入濃度為0.2%的Poloxamer 188、TPGs和Tween-80作為表面活性劑制備木犀草素納米混懸劑,測定粒徑分布及Zeta電位,篩選表面活性劑。研究結果表明,不同種類的表面活性劑會對木犀草素納米混懸劑的粒徑及Zeta電位大小產生一定的影響,使用TPGs作為表面活性劑制備的納米混懸劑粒徑分布最小,Zeta電位較低,因此本研究使用TPGs作為表面活性劑制備木犀草素納米混懸劑。
2.3.4 表面活性劑濃度篩選 固定藥物濃度為30 mg/mL,碾磨介質與混懸劑體積之比為1∶1,碾磨速度為2 500 r/min,碾磨時間為4 h,HPC SL濃度為2%,在處方中分別加入濃度為 0.1、0.15、0.2、0.25、0.3%的TPGs制備木犀草素納米混懸劑,測定粒徑分布及Zeta電位,篩選表面活性劑濃度。研究結果表明,表面活性劑濃度對木犀草素納米混懸劑的粒徑影響不大,但對Zeta電位影響較為顯著,隨著TPGs濃度的增大,Zeta電位升高,因此表面活性劑濃度需要進一步優化。
2.3.5 藥物濃度篩選 固定碾磨介質與混懸劑體積之比為1∶1,碾磨速度為2 500 r/min,碾磨時間為4 h,HPC SL濃度為2%,TPGs濃度為0.2%,藥物濃度分別為 10、20、30、40 mg/mL 制備木犀草素納米混懸劑,測定粒徑分布及Zeta電位,篩選表面活性劑。研究結果表明,藥物濃度對木犀草素納米混懸劑的粒徑具有一定影響,隨著藥物濃度的增大,納米混懸劑的粒徑先減小后增大,而藥物濃度對Zeta電位影響不顯著,因此藥物濃度需要進一步優化。
2.3.6 碾磨介質用量篩選 固定藥物濃度為30mg/mL,碾磨速度為2 500 r/min,碾磨時間為4 h,HPC SL濃度為2%,TPGs濃度為0.2%,分別選擇碾磨介質與混懸劑體積之比為 0.2∶1,0.4∶1,0.6∶1,0.8∶1,1∶1,制備木犀草素納米混懸劑,測定粒徑分布及Zeta電位,篩選碾磨介質用量。研究結果表明,碾磨介質體積對木犀草素納米混懸劑的粒徑具有一定的影響,隨著碾磨介質體積的增加,制備的納米混懸劑粒徑逐漸減小,碾磨介質體積對Zeta電位影響不顯著,因此本研究確定碾磨介質體積與混懸劑體積之比為 1∶1。
2.3.7 碾磨時間篩選 固定藥物濃度為30 mg/mL,碾磨介質與混懸劑體積之比為1∶1,碾磨速度為2 500 r/min,HPC SL濃度為2%,TPGs濃度為0.2%,分別選擇碾磨時間為1、2、4、6 h,制備木犀草素納米混懸劑,測定粒徑分布及Zeta電位,篩選碾磨時間。研究結果表明,碾磨時間對木犀草素納米混懸劑的粒徑產生一定的影響,對Zeta電位影響不顯著,隨著碾磨時間的增加,制備的納米混懸劑粒徑逐漸減小,但是碾磨時間達到6 h,粒徑不再減小,因此確定碾磨時間為4 h。
2.3.8 碾磨速度篩選 固定藥物濃度為30 mg/mL,碾磨介質與混懸劑體積之比為1∶1,碾磨時間為4 h,HPC SL濃度為2%,TPGs濃度為0.2%,分別選擇碾磨速度為 1 000、1 500、2 000、2 500、3 000 r/min,制備木犀草素納米混懸劑,測定粒徑分布及Zeta電位,篩選碾磨速度。研究結果表明,碾磨速度對木犀草素納米混懸劑的粒徑產生一定的影響,對Zeta電位影響不顯著,隨著碾磨速度的增加,制備的納米混懸劑粒徑逐漸減小,但是碾磨速度超過2500r/min,粒徑不再減小,因此確定碾磨速度為2 500 r/min。
2.4 實驗設計
2.4.1 Box-Behnken實驗設計優化 根據單因素實驗篩選結果,以藥物濃度(X1),穩定劑濃度(X2)和表面活性劑濃度(X3)作為考察因素,以木犀草素納米混懸劑的粒徑分布(Y1)布和 Zeta電位(Y2)作為評價指標,通過Box-Behnken實驗設計優化木犀草素納米混懸劑的處方,3個變量水平見表1,生成的15組實驗方案及結果見表2。

表1 Box-Behnken實驗設計中變量及水平Tab.1 Variables and levels of Box-Behnken experiment design
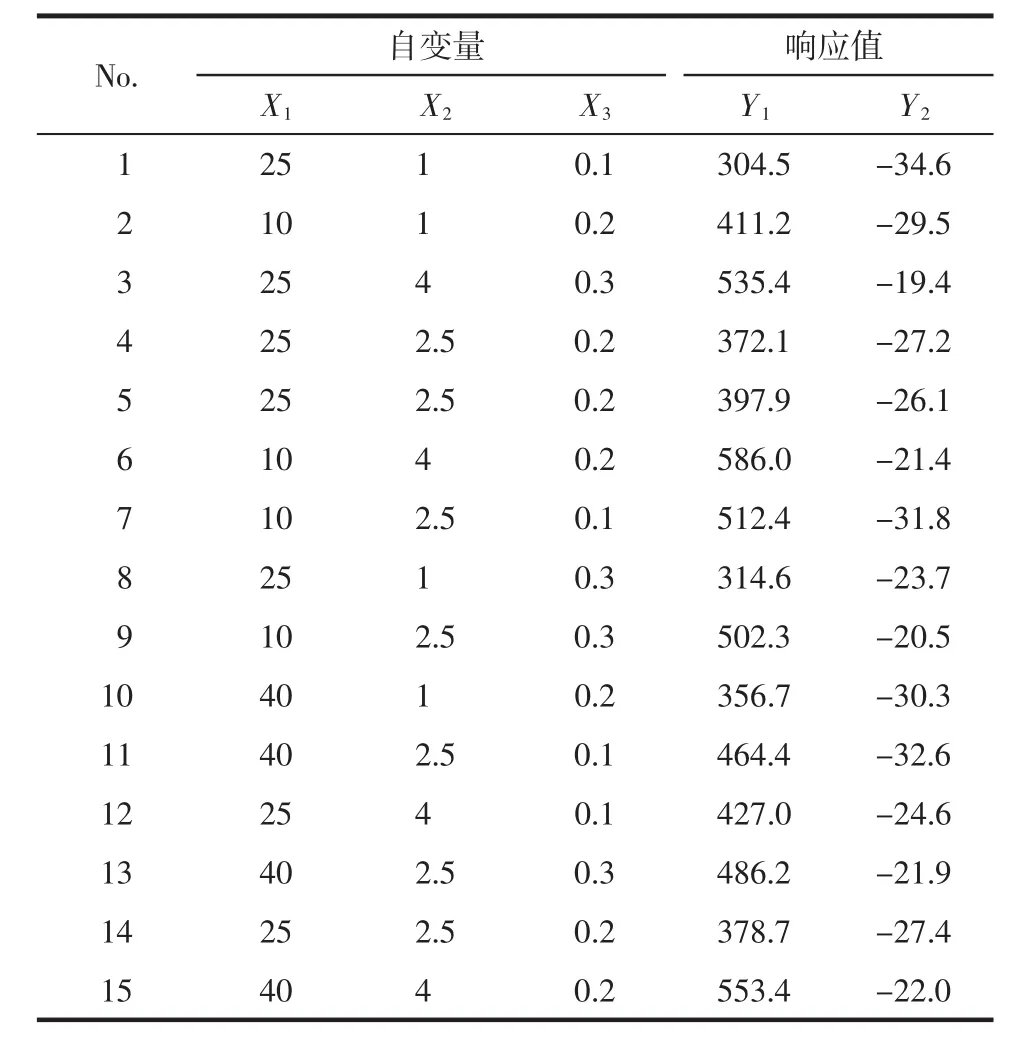
表2 實驗設計及結果Tab.2 The experimental design and results
2.4.2 數據處理及模型擬合 通過“Box-Behnken實驗設計”統計分析實驗結果見表3,兩個模型P值均為 0.000 8,小于 0.05,表明兩個擬合模型均顯著;模型中R2和矯正R2用于估模型方程的可接受性,Y1的 R2值和矯正 R2值分別為 0.982 2 和 0.950 2,Y2的 R2值和矯正 R2值分別為 0.981 4 和 0.947 8,之間差值均小于0.1,說明兩個模型可信度較高;失擬項 P 值分別為 0.254 6 和 0.236 1,均大于 0.05,說明兩個模型的預測值與實際值之間無統計學差異,模型預測性較高。Y1和Y2響應值與3個自變量之間的相關性均符合多元二次方程模型。
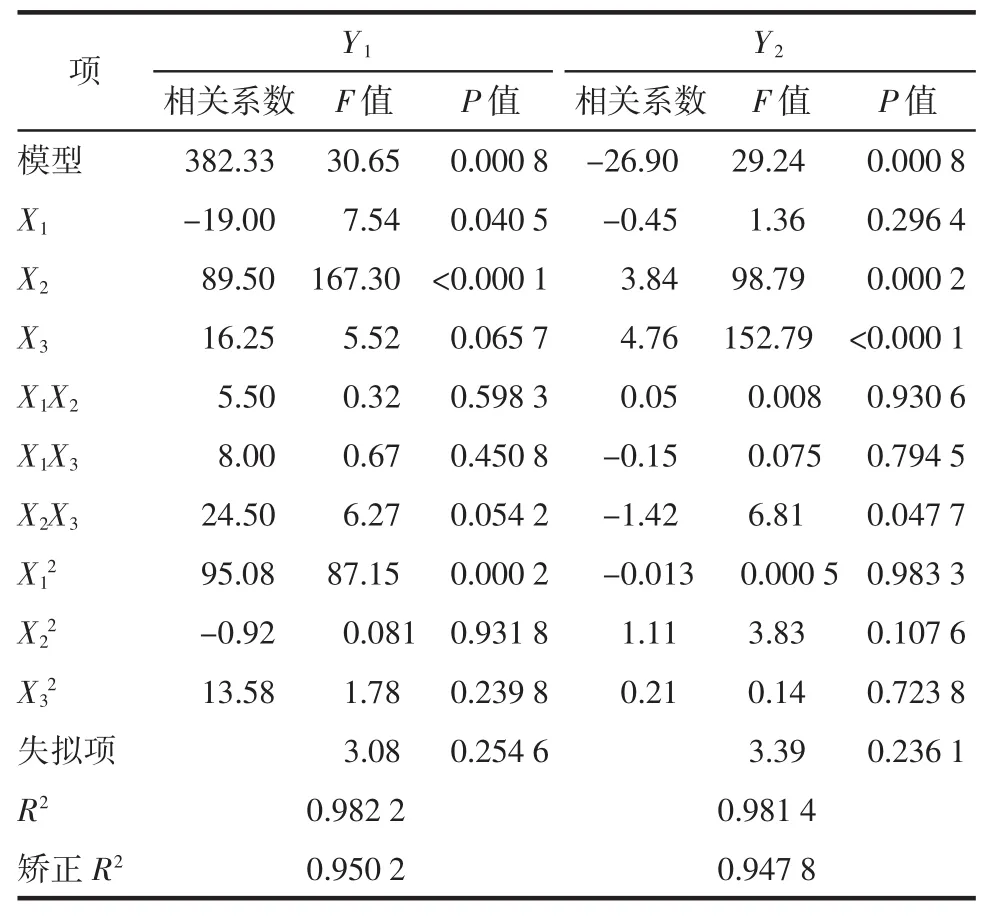
表3 方差分析結果Tab.3 The results of ANOVA
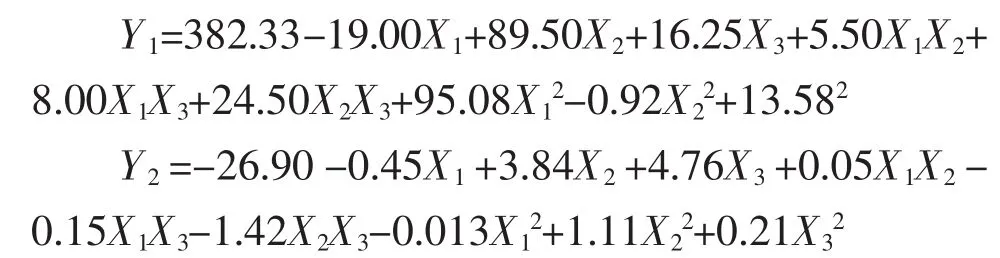
另外,通過方差分析結果可知3個變量與響應值之間的關系,即:X1、X2、X2X3、X12對粒徑分布(Y1)具有顯著影響(P<0.05),而 X2、X3、X2X3對 Zeta 電位(Y2)具有顯著影響(P<0.05)。自變量與響應值之間的關系見圖1、2。
由效應面圖1可知,木犀草素納米混懸劑的粒徑隨著藥物濃度的增加先減小后增大,隨著穩定劑濃度的增加粒徑增大,表面活性劑濃度對粒徑的影響可以忽略不計。
由效應面圖2可知,木犀草素納米混懸劑的Zeta電位隨著穩定劑濃度的增加而升高,也隨著表面活性劑濃度的增加而升高,藥物濃度對Zeta電位的影響可以忽略不計。
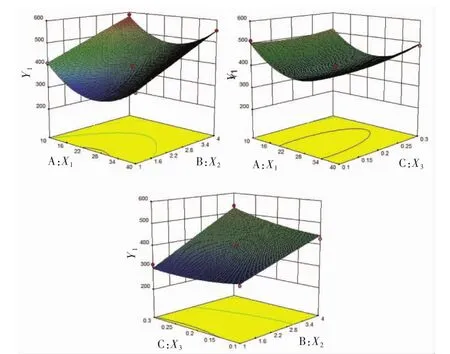
圖1 藥物濃度(X1),穩定劑濃度(X2)、表面活性劑濃度(X3)對木犀草素納米混懸劑的粒徑分布(Y1)的效應面圖Fig.1 Effect of luteolin concentration(X1),stabilizer concentration(X2),surfactant concentration(X3)on the particle size distribution(Y1)of luteolin nanosuspension
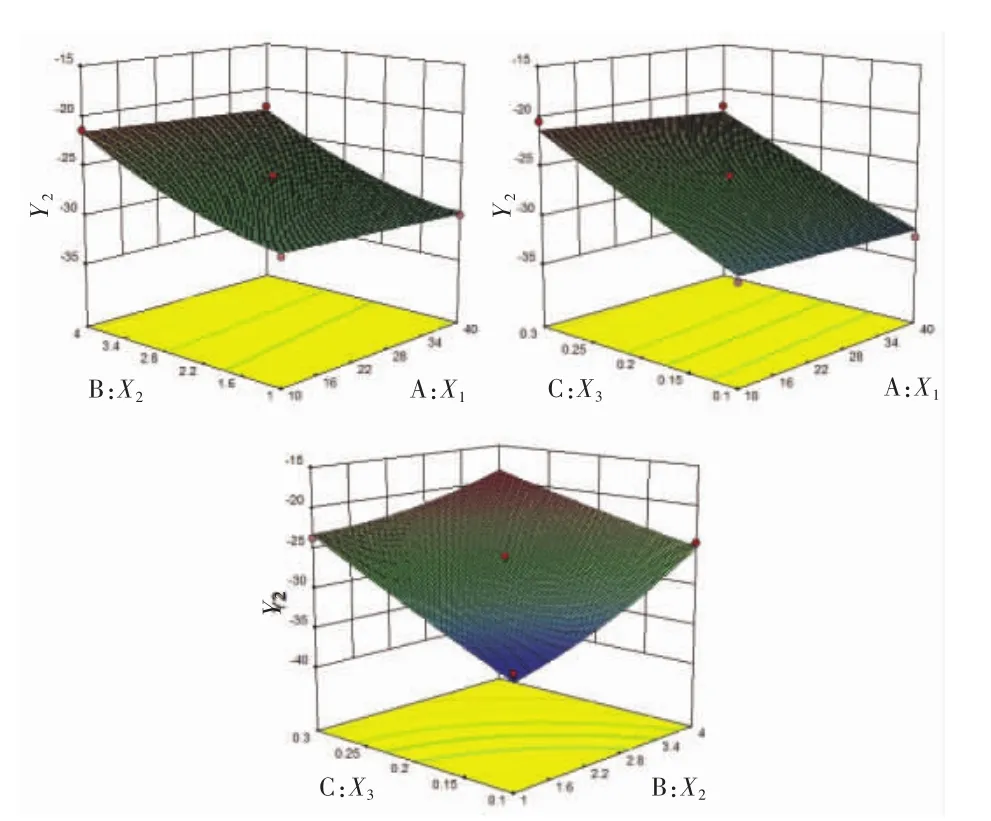
圖2 藥物濃度(X1),穩定劑濃度(X2)、表面活性劑濃度(X3)對木犀草素納米混懸劑的Zeta電位(Y2)的效應面圖Fig.2 Effect of luteolin concentration(X1),stabilizer concentration(X2),surfactant concentration(X3)on Zeta potential(Y2)of luteolin nanosuspension
2.4.3 模型預測與驗證 本研究確定木犀草素納米混懸劑的處方應具有粒徑分布“最小”,Zeta電位“最小”,通過“Box-Behnken實驗設計”優化得到木犀草素納米混懸劑的最優處方組成為:藥物濃度為28.0 mg/mL,穩定劑濃度為1.5 mg/mL,表面活性劑濃度為0.2 mg/mL,軟件預測木犀草素納米混懸劑的粒徑為332.2 nm,Zeta電位為-30.5 mV。根據優化的木犀草素納米混懸劑處方制備3批樣品,經測定粒徑為(324.3±21.6)nm,Zeta電位為(-31.4±0.9)mV,實測值與預測值接近,模型預測可靠性較高。
2.5 微觀形態 取木犀草素納米混懸劑加入少量蒸餾水稀釋,取1滴樣品滴加到載玻片上,揮干水分,噴金,放在掃描電鏡下觀察木犀草素納米混懸劑的微觀形態。掃描電鏡照片顯示木犀草素納米混懸劑呈顆粒狀均勻分布,粒徑大約在100~500 nm之間。見圖3。

圖3 木犀草素納米混懸劑掃描電鏡(×20 000)Fig.3 The scanning electron microscope of luteolin nanosuspension(×20 000)
2.6 體外釋放研究 使用槳法測定木犀草素納米混懸劑與木犀草素原料藥的體外溶出度。溶出介質為0.5%(w/w)吐溫-80磷酸鹽緩沖溶液(pH6.8),體積為500 mL,槳板轉速為50 r/min,水浴溫度為37℃。開啟溶出儀,用移液器精密移取0.5 mL木犀草素納米混懸劑加入到溶出杯中,分別在5、10、20、30、45、60 min取樣 5 mL 溶出介質,20 000 r/min高速離心,取1 mL上清液經適當稀釋,使用HPLC法[14]測定藥物含量;另取15 mg木犀草素原料藥加入到溶出杯中,按照上述方法操作,測定藥物含量。通過繪制時間-溶出度曲線比較木犀草素納米混懸劑與木犀草素原料藥的溶出度。見圖4。
由木犀草素納米混懸劑與木犀草素原料藥溶出曲線可知,木犀草素原料藥在10 min累積溶出度為18.3%,30 min累積溶出度為34.4%,60 min累積溶出度為42.2%,體外累積溶出度數據經威布爾方程擬合為:lnln[1/(1-C(t))]=0.6054ln(t)-2.994 9(R2=0.988 9),經計算體外累積溶出度50%的時間為77.6 min,累積溶出度63.2%的時間為89.6 min;而木犀草素納米混懸劑在10 min溶出度為76.4%,30 min溶出度為96.3%,60 min溶出度98.7%,體外累積溶出度數據經威布爾方程擬合為:lnln[1/(1-C(t))]=0.771 8ln(t)-1.573 3(R2=0.971),經計算體外累積溶出度50%的時間為4.8 min,累積溶出度63.2%的時間為7.7 min,表明木犀草素納米混懸劑能夠顯著提高藥物溶出度。
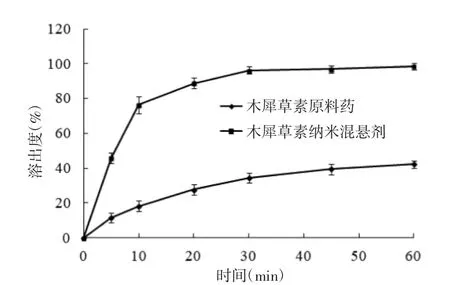
圖4 木犀草素納米混懸劑與木犀草素原料藥體外溶出曲線(n=6)Fig.4 In vitro release curve of luteolin nanosuspension and luteolin(n=6)
3 討論
目前,藥物納米粒子的制備技術從原理上可分為“自上而下”(Top-down)和“自下而上”(Bottomup)兩種制備技術[15]。“自下而上”分為反溶劑沉淀法和化學反應沉淀法,“自上而下”分為介質碾磨法、高壓均質法和微射流法,由于介質碾磨法在納米混懸劑制劑生產中具有高度的可重現性、可避免碾磨腔體遞質污染、易于規模放大,因此本研究選用介質碾磨法制備木犀草素納米混懸劑。
納米混懸劑屬于熱力學不穩定系統,在處方中需要添加穩定劑以降低液-固界面張力,提高體系的穩定性。本研究在木犀草素納米混懸劑的處方中加入HPC SL和TPGs作為穩定劑,HPC SL可以吸附到納米混懸劑表面,形成一層薄膜,阻礙顆粒之間的聚集,起到空間位阻作用,而表面活性劑TPGs可降低了藥物與水之間的界面張力,潤濕藥物表面,通過穩定劑與表面活性物質的雙重作用提高了木犀草素納米混懸劑的穩定性。
