高尿酸背景下P2X7R的Arg 307>Gln位點基因型的功能研究
2020-07-13 03:51:14李曼云陶金輝潘顯陽厲小梅李向培
安徽醫科大學學報 2020年6期
李曼云,陶金輝,方 璇,馬 艷,潘顯陽,厲小梅,李向培
痛風是一種由高尿酸血癥和尿酸單鈉鹽(monosodium urate,MSU)晶體沉積引起的炎癥性疾病。目前有研究[1]表明,細胞外三磷酸腺苷(adenosine triphosphate, ATP)的變化可激活嘌呤受體P2X配體門控離子通道7(purinergic receptor P2X ligand-gated ion channel 7, P2X7R)信號通路,協同MSU晶體刺激產生核苷酸結合寡聚化結構域樣受體3[oligomerization domain (NOD)-like receptor family, pyrin domain containing 3,NLRP3]炎癥小體并激活半胱氨酸天冬氨酸蛋白酶-1 (Caspase-1),使白細胞介素(interleukin,IL)-1β分泌量增加,導致痛風的發作。因此,ATP的顯著變化是導致急性痛風性關節炎發生的關鍵。另外,臨床有部分高尿酸血癥患者一生從未發作痛風,可能與P2X7R多態性相關。因此,P2X7R可能是急性痛風性關節炎發作的一個關鍵調節因子。人類P2RX7基因上存在許多單核苷酸多態性(single nucleotide polymorphism,SNP)位點。既往有研究[2-3]報道,攜帶人P2RX7基因Arg 307>Gln突變體的細胞使P2X7R功能降低甚至缺失。現擬探討在人急性白血病單核細胞株 (human leukemic monocyte,THP-1)源性巨噬細胞中,通過建立高尿酸鹽的背景,并分別轉染野生型、突變體型Arg 307>Gln和空病毒型THP-1細胞系,比較3種P2X7R功能的差異。
1 材料與方法
1.1 實驗材料人急性白血病單核細胞株THP-1購自中國科學院上海細胞生物學研究所;胎牛血清與RPMI-1640 培養基均購自美國Gibco 公司;綠色熒光蛋白(green fluorescent protein,GFP)-慢病毒載體(lentivirus,LVs)購自上海漢恒生物科技有限公司;微量RNA提取試劑盒、 cDNA 逆轉錄試劑盒及 SYBR?Green PCR試劑盒均購自德國QIAGEN公司;P2X7R、 IL-1β、NLRP3、 凋亡相關斑點樣蛋白含半胱氨酸天冬氨酸-1蛋白酶結構域(apoptosis-associated speck-like protein containing a caspase-1 recruitment domain,ASC)、Caspase-1及β-actin PCR引物均購自上海生工生物科技有限公司;IL-1β試劑盒購自美國R&D Systems公司。
1.2 THP-1細胞的培養將THP-1細胞培養于10%胎牛血清的RPMI-1640培養基中,5% CO2、37 ℃溫箱中培養,密切觀察THP-1細胞,培養期間每3 d更換1次培養基。
1.3 慢病毒搭載GFP熒光轉染THP-1細胞并篩選目的基因取1×105個THP-1細胞,接種于48孔板培養,加入完全培養基培養過夜。第2天細胞融合達40% ~50%,加入GFP-LVs,同時加入聚凝胺后搖勻,放入37 ℃溫箱中培養。轉染72 h后,熒光倒置顯微鏡DMI3000B(德國 Leica公司)下觀察慢病毒轉染情況。轉染成功后再次傳代,并在熒光倒置顯微鏡下觀察熒光是否衰減。
1.4 實驗細胞分組及功能實驗將轉染過的THP-1細胞先用丙二醇甲醚醋酸酯 (phorbol-12-myristate-13-acetate,PMA)刺激3 h后,在野生型、突變體型Arg 307>Gln和空病毒型中各設置3組:MSU(M組)、MSU+ATP(MA組)、對照組(C組),M組、MA組中加入MSU孵育6 h后, MA組中加入ATP孵育 1 h,分別收取3組中轉染后THP-1細胞上清液和細胞。
1.5 ELISA檢測細胞上清液中IL-1β各標本細胞上清液凍存于-80 ℃,實驗時各標本置于4 ℃緩慢解凍,1 000 r/min、4 ℃離心15 min,根據說明書檢測上清液中IL-1β表達水平。
1.6 qRT-PCR檢測mRNA表達水平TRIzol(美國Invitrogen公司)溶液提取細胞總RNA,-80 ℃冰箱保存,最后集中進行定量測定。使用ABI7500全自動實時定量PCR儀進行檢測,以GAPDH 為內參。反應條件: 95 ℃、2 min,95 ℃、5 s,60 ℃、34 s,40個循環。融解曲線分析: 溫度60~95 ℃ ,每個樣本重復3次,以保證結果的準確性。采用 ΔCt 和 2-ΔΔCt法計算目的基因的相對表達量。
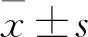
2 結果
2.1 在MSU背景下,突變體型Arg 307>Gln降低了ATP刺激P2X7R分泌IL-1β的能力MA組中野生型、突變體型Arg 307>Gln和空病毒型THP-1細胞上清液和細胞中IL-1β表達水平差異均有統計學意義(F=4.606,P=0.035 2;F=9.063,P=0.001 14);其中,與野生型相比,突變體型Arg 307>Gln上清液中IL-1β表達水平降低(P=0.016 4)(見圖1A),細胞中IL-1β mRNA表達水平也降低(P<0.001)(見圖1B)。然而,野生型和突變體型Arg 307>Gln與空病毒型相比THP-1細胞上清液和細胞中IL-1β表達水平均基本無差異(上清液:P=0.120 8、P=0.597 2;細胞:P=0.119 9、P=0.226 1)。C組和M組的野生型、突變體型Arg 307>Gln和空病毒型上清液和細胞之間差異均無統計學意義(上清液C組:F=0.891 5,P=0.458 1;上清液M組:F=2.375,P=0.138 9;細胞C組:F=4.794,P=0.057 0;細胞M組:F=1.399,P=0.301 2)。見圖1。

圖1 3種THP-1細胞上清液及細胞中IL-1β表達水平比較
A:上清液中IL-1β濃度; B:細胞中IL-1β mRNA;與野生型比較:*P<0.05,***P<0.001
2.2 在MSU背景下,突變體型Arg 307>Gln抑制了ATP刺激P2X7R對NLRP3的調控MA組中,野生型、突變體型Arg 307>Gln和空病毒型THP-1細胞中NLRP3 mRNA的表達差異均無統計學意義(F=7.958;P=0.020 5);其中,與野生型比較,突變體型中NLRP3 mRNA表達水平降低(P=0.027 9)。然而,野生型和突變體型Arg 307>Gln與空病毒型相比NLRP3 mRNA表達水平差異均無統計學意義(P=0.085 9、P=0.491 5)。C組和M組中野生型、突變體型Arg 307>Gln和空病毒型之間NLRP3 mRNA的表達水平均亦無差異(分別是F=3.906,P=0.082 0;F=0.339 8,P=0.721 7)。見圖2。

圖2 3種THP-1細胞中NLRP3 mRNA表達水平比較
2.3 MSU背景下,野生型和突變體型Arg 307>Gln THP-1細胞中ASC表達均顯著增高,突變體型Arg 307>Gln不影響ATP刺激P2X7R對ASC的調控MA組中,野生型、突變體型Arg 307>Gln和空病毒型THP-1細胞中ASC mRNA的表達水平存在差異(F=14.22,P=0.005 3),野生型和突變體型Arg 307>Gln組的ASC mRNA表達水平升高,但2組間ASC mRNA表達水平均無差異(P=0.169 4)。然而,野生型和突變體型Arg 307>Gln與空病毒型相比,ASC mRNA表達水平均存在差異(P=0.002 2、P=0.028 3)。C組和M組中野生型、突變體型Arg 307>Gln和空病毒型三者之間ASC mRNA的表達水平差異均無統計學意義(F=1.097,P=0.379 2;F=4.761,P=0.057 8)。

圖3 3種THP-1細胞中ASC mRNA表達水平比較
2.4 在MSU背景下,突變體型Arg 307>Gln THP-1細胞不影響ATP刺激P2X7R對Caspase-1的調控C組、M組和MA組中,野生型、突變體型Arg 307>Gln和空病毒型THP-1細胞中Caspase-1 mRNA表達水平差異均無統計學意義(F=0.591 5,P=0.582 8;F=2.445,P=0.167 2;F=0.903 8,P=0.453 8)。

圖4 3種THP-1細胞中Caspase-1 mRNA表達水平比較
3 討論
痛風作為一種自身炎癥性疾病, IL-1β在MSU誘導的炎癥通路中起著不可或缺的作用[4]。IL-1β的產生有3個步驟:IL-1β前體(pro-IL-1β)蛋白的產生;切割pro-IL-1β產生活性IL-1β蛋白;將IL-1β釋放到細胞外環境中。其中, pro-IL-1β的處理,涉及到Caspase-1的激活,也是炎癥小體活化的基本特征。
急性痛風性關節炎與MSU晶體形成密切相關。已有文獻[5]證明,在體內MSU可被膜結合受體(如Toll樣受體,TLRs)和細胞內模式識別受體(如NOD樣受體)識別。THP-1源性單核細胞在PMA的作用下分化為THP-1源性巨噬細胞并通過Toll 2樣受體(Toll-like receptor 2,TLR2)和TLR4識別MSU晶體破壞溶酶體,釋放到胞質中的溶酶體組織蛋白酶B可結合NLRP3并激活NLRP3炎癥小體[6]。在NOD樣受體(nucleotide-binding oligomerization domain-like receptors,NLRs)的作用中,NLRP3可以通過其富含亮氨酸的重復序列結構域(leucine rich repeat,LRR)識別并結合MSU晶體參與痛風發病。目前認為PMA通過激活NF-κB 通路誘導 pro-IL-1β、半胱氨酸天冬氨酸蛋白酶-1前體(pro-caspase-1)的表達,在NLRP3炎癥小體的作用下使pro-caspase-1活化成為Caspase-1,促使 pro-IL-1β分化成為成熟IL-1β[7]。
既往Fettelschoss et al[8]用NLRP3激活劑即脂多糖刺激單核細胞系,后用ATP刺激。而Jhang et al[6]用PMA先刺激THP-1細胞分化為巨噬細胞系,再用MSU晶體激活下游信號通路,使IL-1β的分泌量顯著增加。研究[6]發現pro-IL-1β水平上調,但NLRP3炎癥小體和pro-caspase-1的表達水平不受影響。本研究中,使用ATP分別干預經PMA誘導并轉染過野生型、突變體型Arg 307> Gln和空病毒型的THP-1源性巨噬細胞,比較它們分泌IL-1β的能力以及ATP-P2X7R-IL-1β信號通路上3個因子的表達水平。在MSU晶體建立的高尿酸背景下,由ATP孵育的野生型THP-1細胞上清液IL-1β和細胞中IL-1β、NLRP3的mRNA表達水平均顯著高于突變體型。提示突變體型Arg 307> Gln可能影響了P2X7R功能,使ATP誘導的ATP-P2X7R-IL-1β通路分泌IL-1β的能力明顯降低,從而可能降低了炎癥的發生及痛風的發作。
NLR家族包含2種炎癥小體,分別為NLRP3和黑色素瘤缺乏因子2(absent in melanoma 2, AIM2)。當識別特異性激動劑后,NLRP3和AIM2可分別形成炎癥小體。而凋亡相關斑點樣蛋白含半胱氨酸天冬氨酸-1蛋白酶結構域(apoptosis-associated speck-like protein containing a caspase-1 recruitment domain ,ASC)是一種銜接NLRP3和AIM2的銜接蛋白,它們相互作用。一方面,ASC可募集pro-caspase-1到NLRP3炎癥小體復合物中,另一方面NLRP3和AIM2可誘導幾乎所有的ASC分子聚合,形成Caspase-1活化平臺—ASC斑點[9],從而誘導了Caspase-1的活化和成熟[10]。在本研究中,當MSU或ATP作為激動劑作用于野生型和突變體型THP-1細胞時, MA組野生型ASC mRNA表達水平高于突變體型,但差異無統計學意義,推測與ASC斑點形成后大部分被釋放到細胞外,相鄰巨噬細胞通過吞噬作用攝取ASC斑點導致部分ASC損失有關[10-11]。
近年來,Keller et al[12]提出Caspase-1可激活其他底物,調控大量細胞因子和應激信號的分泌。Caspase-1還可以切割IL-1β的前體分子,使pro-IL-1β成為有活性成熟的IL-1β蛋白并在巨噬細胞中分泌出來[4]。由于Caspase-1可催化失活的酶原,通常在催化過程中自身被蛋白酶水解。本研究中, 各組Caspase-1蛋白的mRNA表達水平基本無差異,推測與其自身被蛋白酶水解有關[13]。
猜你喜歡
音樂探索(2022年2期)2022-05-30 21:01:37
美與時代·美術學刊(2022年3期)2022-04-27 01:18:15
火花(2019年12期)2019-12-26 01:00:28
小天使·一年級語數英綜合(2019年8期)2019-08-27 02:23:00
人大建設(2019年12期)2019-05-21 02:55:32
中國特種設備安全(2018年11期)2019-01-08 02:08:32
小學科學(學生版)(2018年7期)2018-08-13 09:33:04
學苑創造·A版(2015年11期)2016-01-14 09:03:27
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:46
山東女子學院學報(2014年6期)2014-03-01 02:24:55
