黃芪多糖對C3H10T1/2細胞棕色脂肪分化中長鏈非編碼RNA表達譜的影響
2020-09-11 07:40:26張世和曹宇鑫鄧步皓高旭陽趙俊星
激光生物學報 2020年4期
張世和,曹宇鑫,鄧步皓,高旭陽,趙俊星
(山西農業大學動物科技學院,太谷 030800)
黃芪多糖(astragalus polysacharin,APS)提取自中藥材黃芪的根部,作為黃芪最重要的天然有效成分之一發揮作用。隨著人們對多糖研究的加深,其在抗氧化、抗炎、免疫調節、抗腫瘤、降血脂、降血糖等多方面的生物活性與功能也隨之被發現[1-4]。APS也因其能促進3T3-L1前脂肪細胞的增殖和分化[5]以及抑制營養消化和吸收酶的活性來治療肥胖癥而備受關注[6]。近年來的研究多將APS運用于2型糖尿病、肥胖和代謝綜合征等代謝相關性疾病的防治上。
棕色脂肪(brown adipose tissue,BAT)是哺乳動物發熱生熱的關鍵部位,其產生的熱量對于初生哺乳動物在寒冷環境中的生存及冬眠動物的體溫維持至關重要。BAT由于含有大量的線粒體以及解耦聯蛋白 1(uncoupling protein 1,UCP1)成為非寒顫性熱發生的主要組織,從而增加能量消耗[7]。近年來的研究表明,BAT能通過攝入甘油三酸酯來加速血漿脂質的清除,從而改善血脂異常和胰島素抵抗[8]。在人和小鼠中的研究均發現,BAT可積極利用線粒體中的支鏈氨基酸進行熱發生,進而控制肥胖的發生[9-10]。因此,通過激活BAT來增加能量消耗已成為一種安全的預防和控制肥胖的方法。
長鏈非編碼 RNAs(long non-coding RNAs,lncRNAs)參與不同的生物學過程[11],并在器官發育過程中發揮著關鍵作用,包括細胞增殖、細胞分化、細胞周期的調節、凋亡和衰老[12]。近年來的研究發現,lncRNAs具有調節脂肪形成的能力[13-14],并鑒定出多個與棕色脂肪發育相關的lncRNAs。lncRNA-Blnc能與早期B細胞因子2(recombinant early B-cell factor 2,EBF2)形成核糖核蛋白復合體來增強EBF2的表達,并通過調節產熱基因來促進小鼠米色和棕色脂肪細胞的分化[15]。lncBATE1能維持棕色脂肪細胞核心標記基因的表達,抑制白色脂肪標記基因,且能與核異質核糖核蛋白U(heterogeneous nuclear ribonucleoprotein U,hnRNP U)相互作用以促進米色和棕色脂肪的形成[16]。
本研究以小鼠間充質干細胞C3H10T1/2為研究對象,在體外培養的過程中添加APS,收集樣品并進行轉錄組測序分析,旨在揭示APS促進棕色脂肪細胞C3H10T1/2分化的作用機理,并闡明lncRNAs在棕色脂肪細胞分化中的功能。
1 材料與方法
1.1 細胞和主要試劑
C3H10T1/2細胞購買自美國模式培養物集存庫(American type culture collection,ATCC);達爾伯克改良伊格爾低糖培養液(Dulbecco’s modified eagle medium,DMEM,SH30021.01)購自于 Hyclone公司;雙抗(青鏈霉素,10378016)與胎牛血清(10091155)購自于Gibco公司;三碘甲腺原氨酸(3′-triiodothyronine,T3)、吲哚美辛、胰島素(I5500)、地塞米松(dexamethasone,Dex,D4902)和 3-異丁基 -1-甲基黃嘌呤(3-isobutyl-1-methyl-7H-xanthine,IBMX,I7018)購自于美國Sigma公司;PrimeScriptTMRT Master Mix(R047A)與 SYBR?Premix ExTaqTMII65(R820A)購于日本TaKaRa公司。
1.2 C3H10T1/2細胞的培養和棕色化誘導
C3H10T1/2細胞種植在生長培養基(含2%雙抗、10%胎牛血清的低糖DMEM)中,在37℃、5%CO2的細胞培養箱中培養。待細胞鋪滿培養皿后,分別使用未添加和添加0.4 g/L APS的分化培養基(0.5 mmol/L IBMX、5 μmol/L Dex、1 nmol/L T3、0.125 mmol/L 吲哚美辛和10 g/L胰島素溶于含有2%雙抗的10%胎牛血清的DMEM培養基中)誘導分化,分化1.5 d后取樣用于后續試驗。
1.3 cDNA文庫的構建和測序
以未經APS處理的棕色脂肪細胞為對照組,經0.4 g/L APS處理為試驗組,每組做3個生物學重復,構建6個cDNA文庫進行測序。上機前樣品提取總RNA后,將rRNAs去除以保留mRNAs和ncRNAs。使用片段緩沖液將得到的mRNAs和ncRNAs片段化為短片段,并用隨機引物將其逆轉錄為cDNA第一鏈。接著加入緩沖液、dNTPs(dUTP代替dTTP)、核糖核酸酶 H(ribonuclease H,RNase H)和 DNA polymerase I合成cDNA第二鏈。通過使用QiaQuick PCR試劑盒純化cDNA片段進行末端修復,并加堿基A和Illumina測序接頭。然后使用尿嘧啶-N-糖基化酶(Uracil-N-glycosylase,UNG)降解第二鏈 cDNA。通過瓊脂糖凝膠電泳選擇降解產物的大小,進行PCR擴增。最后通過Illumina HiSeqTM4000平臺對文庫制備物進行測序。
1.4 測序數據過濾
利用 FastQC(v0.11.4)(http://www.bioinformatics.babraham.ac.uk...jects/fastqc/)分析了reads的堿基組成、質量分布以及GC和AT的堿基含量。通過刪除全為A堿基、具有10%以上N堿基和質量值(Q≤20)超過整條reads 50%的低質量reads,來得到高質量的reads用于后續的信息分析。
1.5 比對分析及重構轉錄本
使用reads比對工具bowtie2[17](2.2.8)和比對軟件Tophat2[18](2.1.1)將高質量的reads比對到該物種的核糖體中(錯配數:0),去掉比對上的reads后再比對到物種的參考基因組上。保留比對上的數據,使用Cufflinks[19]來重構轉錄本,根據組裝出來的轉錄本在參考基因組上的位置以及篩選標準(轉錄本長度≥200 bp且exon數目≥2)篩選新的轉錄本,從而得到樣本已知的和新的轉錄本。
1.6 新lncRNAs預測
對篩選出來的新轉錄本進行lncRNAs預測。使用CPC[20]和CNCI[21]2個軟件預測其是否具有編碼蛋白的能力,同時到蛋白數據庫SwissProt中進行比對,取預測無編碼能力且在蛋白數據庫中無法比對上的轉錄本作為新lncRNA。
1.7 差異表達基因的分析
使用edgeR軟件(http://www.r-project.org/)對組間lncRNAs和mRNAs的表達量進行差異分析,利用P值與log2FC來篩選差異mRNAs轉錄本,篩選條件為P<0.05且|log2FC|>1。
1.8 GO功能注釋和KEGG通路分析
為了解lncRNAs靶基因的功能作用,我們使用基因本體(gene ontology,GO)和京都基因與基因組百科全書(Kyoto Encyclopedia of Genes and Genomes,KEGG)富集分析對其進行了研究。GO總共有3個本體,分別描述轉錄本的分子功能、細胞組分和參與的生物過程。KEGG是分析基因在細胞中的代謝途徑的主要公共數據庫[22],尤其適用于分析基因組測序和其他高通量技術,從而得到大規模的數據。
1.9 通過Real-PCR驗證基因和lncRNAs的表達
使用Trizol?Reagent試劑盒提取總RNA,提取后用1%瓊脂糖凝膠電泳檢測其是否降解,并用NanodropND-1000進行濃度與完整性檢測,選取OD260nm/OD280nm為1.9~2.0者用于后續試驗。RNA經DNA酶處理后,取1 μg進行cDNA反轉錄,并進行qRT-PCR分析。擴增程序為:95℃預變性20 s;95℃變性20 s,60℃退火及延伸20 s,36個循環。結果根據2-ΔΔCT法計算,18S rRNA基因為內參基因,引物序列見表1。
2 結果與分析
2.1 數據的來源
從Illumina HiSeqTM4000平臺的每個文本庫中平均產生了15 177 607 700個清潔數據,在去除不符合的堿基比例和低質量的reads后,從每個庫中平均獲得了14 831 667 631個高質量的reads。其中,測序準確率為99.9%的堿基數目占比可以達到95.19%~96.34%(表2)。在去除比對上的核糖體reads后,將其比對到小鼠的參考基因組上,有87.76%~90.65%的reads被成功定位到小鼠的參考基因組上(表2)。使用Cufflinks根據比對結果來重構轉錄本,共得到了 13 450個 lncRNAs和 57 776個 mRNAs。使用CPC、CNCI及蛋白數據庫SwissProt等工具評估新轉錄本的蛋白質編碼潛力,并取結果相交項作為最終結果,我們預測出了586個新lncRNAs轉錄本(圖 1a)。

表1 引物序列Tab.1 Primer sequences for real-time qPCR
2.2 lncRNAs數據的分布和質量分析
根據lncRNAs在基因組上相對于蛋白編碼基因的位置,我們將lncRNAs分為5種類型,其中有13 319個為基因間區型lncRNAs,16個為雙向型lncRNAs,39個為反義 lncRNAs,63 個為正義 lncRNAs,13個為其他類型的 lncRNAs(圖 1b),基因間區lncRNAs是最常見的lncRNAs類型,其比例也最大。在所有的lncRNAs中,大多數lncRNAs具有2個外顯子(最多有27個外顯子,平均3.10個外顯子),顯著低于蛋白質編碼基因(最多有347個外顯子,平均9.51個外顯子)(圖1c)。總體而言,lncRNAs的平均長度為1 251.84 bp,小于mRNAs的2 615.63 bp,lncRNAs和mRNAs長度的分布是一致的,相對較長的mRNAs轉錄物數量高于lncRNAs(圖1d),轉錄本的平均表達值計算方法為每100萬map上的reads中map到轉錄本上的每1 000個堿基上的fragment數目(fragments per kilobase of transcript per million mapped reads,FPKM),FPKM 小提琴圖表明(圖 1e),mRNAs的FPKM值為6.35,lncRNAs的FPKM值為1.75,mRNAs相對表達水平高于lncRNAs。
2.3 成對重復性檢驗
為獲得對試驗結果可靠性和操作穩定性的評估,我們對2次生物學平行試驗的結果進行了相關性分析。lncRNAs的分析結果顯示,樣本間皮爾森相關系數(pearson correlation coefficient,PCC)全部大于0.990 0,最高可達到0.999 7。 mRNAs分析結果顯示,樣本間PCC全部大于0.990 0,最高為0.999 0,說明測序具有可重復性,試驗結果的可靠性和操作的穩定性高(圖2)。
2.4 組間差異表達基因分析
依據lncRNAs和mRNAs表達的火山圖,我們發現了153個差異表達的lncRNAs(differentially expressed lncRNAs,DElncRNAs),其中有 76 個上調,77個下調(圖3a、3c),同時也發現了1 238個差異表達的 mRNAs(differentially expressed mRNAs,DEGs),其中有590個上調,648個下調(圖3b、3c)。隨后我們對這些DEGs進行了GO功能注釋和KEGG分析,GO功能注釋結果顯示(圖3d),DEGs富集在了53個條目中,在3個本體的數量分布為:參與的生物過程24個、分子功能10個及細胞組分19個。參與的生物過程本體的條目主要有代謝過程、生長過程、細胞過程、生殖過程、生物過程調控等,其中細胞過程富集基因最多,有734個;分子功能本體的條目有結合、轉錄因子活性、蛋白質結合、核酸結合轉錄因子活性、分子功能調節劑、抗氧化活性等,其中結合功能富集基因最多,有645個;細胞組分本體的條目有細胞成分、突觸、核苷酸、細胞外基質、細胞膜部分等,其中細胞成分富集基因最多,有753個。KEGG分析顯示(圖3e),這些基因富集在了343條通路中,根據富集顯著性進行排序,排名前20的通路為:系統性紅斑狼瘡、酗酒、賴氨酸降解、癌癥中的轉錄失調、皮質醇合成與分泌、破骨細胞分化、緊密連接、腫瘤壞死因子(tumor necrosis factor,TNF)信號通路、醛固酮合成與分泌、NOD樣受體(nucleotide binding oligomerization domain-like receptors,NLR)信號通路、庫欣綜合征、甲狀旁腺激素的合成分泌及作用、基底切除修復術、脂肪酸代謝、檸檬酸循環、局灶性粘連、人類嗜T細胞病毒I型(human T-cell leukemia virus-I,HTLV-I)感染、促性腺激素釋放激素(gonadotropin-releasing hormone,GnRH)信號通路、環磷酸鳥苷酸-蛋白激酶G(cyclic guanosine monophosphate-protein kinase G,cGMP-PKG)信號通路和代謝途徑。其中TNF信號通路、脂肪酸代謝和檸檬酸循環等通路能積極參與BAT細胞分化的過程。
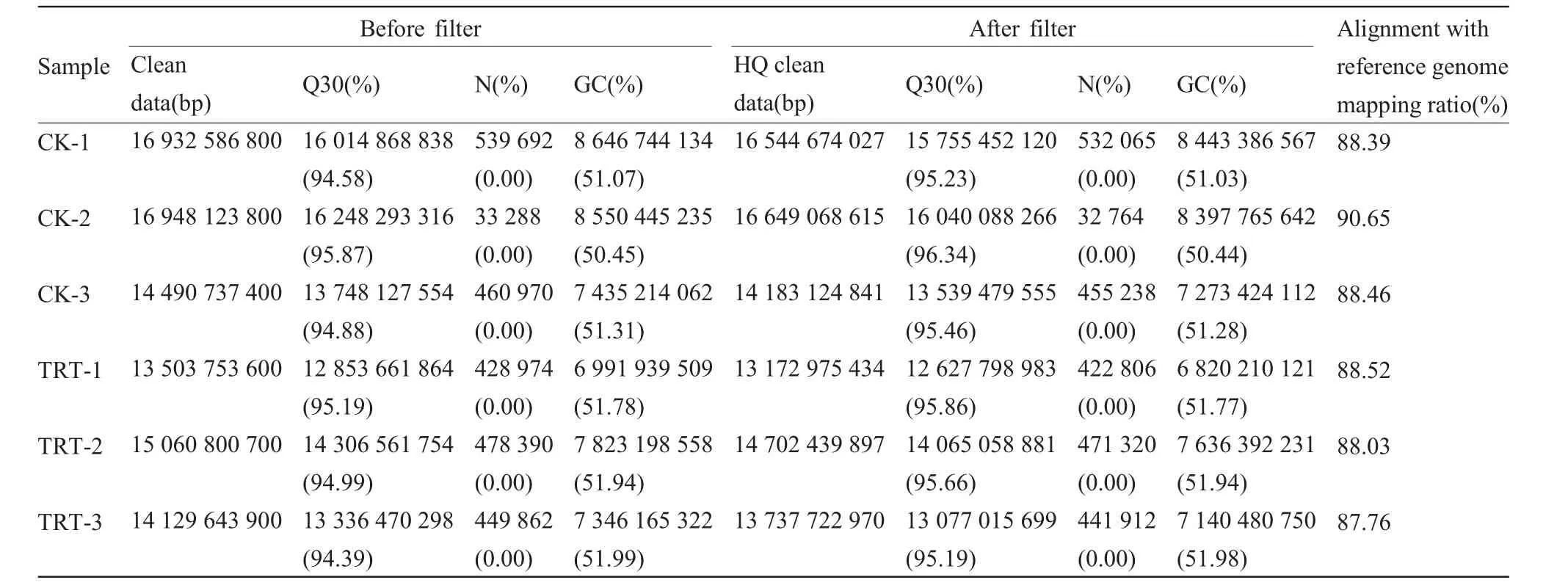
表2 過濾前后堿基信息及比對分析統計表Tab.2 Base information before and after filtering and comparison analysis statistics table

圖1 新lncRNAs轉錄本和數據質量分析Fig.1 New lncRNAs transcript and data quality analysis
2.5 lncRNAs順式調控靶基因的預測

圖2 成對重復性檢驗Fig.2 Pairwise repeatability test
以lncRNAs的上游或者下游10 kb為界定線,我們預測到了108個差異表達lncRNAs具有靶基因。靶基因GO富集分析顯示,其顯著富集的條目有32個,分別為參與的生物過程16個,細胞組分7個,分子功能9個。參與的生物過程中富集的條目有代謝過程、發育過程、生物調節、細胞過程等,其中細胞過程富集基因最多,有66個。細胞組分中富集的條目有細胞器、細胞器部分、細胞部分、細胞等,其中細胞部分和細胞器富集基因最多,都有66個。分子功能中富集的條目有核酸結合轉錄因子活性、結合、結構分子活動、催化活性,其中結合功能富集基因最多,有54個(圖4a)。KEGG分析結果顯示,靶基因富集在了66條通路中。根據KEGG氣泡圖可知,富集顯著性排名前20的通路為:安非他明成癮、胰高血糖素信號途徑、醛固酮合成與分泌、胰島素分泌、多巴胺能突觸、心肌細胞腎上腺素能信號、可卡因成癮、甲狀腺激素合成、HTLV-I感染、單純皰疹感染、低氧誘導因子-1(hypoxia inducible factor-1,HIF-1)信號通路、絲裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)信號通路、哺乳動物壽命調節途徑、TNF信號途徑、雌激素信號途徑、炎癥介質對瞬時受體電位(transient receptor potential,TRP)通道的調節、腸免疫網絡用于免疫球蛋白 A(immunoglobulin A,IgA)的產生、原發性免疫缺陷、乙型肝炎和甲型流感(圖4b)。
2.6 lncRNAs共表達靶基因的預測

圖3 組間差異表達基因分析Fig.3 Analysis of differentially expressed genes between groups
lncRNAs共表達靶基因的GO分析結果顯示(圖5a),預測到的靶基因富集在了33個條目中。參與生物過程本體中的條目有單生物過程、細胞過程、代謝過程、對刺激的反應等,其中細胞過程富集基因最多,有2 244個。在分子功能中的主要條目有結合、分子功能調節劑、核酸結合轉錄因子活性、結構分子活性、轉錄因子活性、蛋白質結合等,其中結合富集基因最多,有1 491個。在細胞組分中的主要條目有細胞外區域、細胞外基質、細胞、細胞器、細胞器部分等,其中在細胞部分富集基因最多,有2113個。
KEGG分析結果顯示,共表達基因主要參與290條代謝通路,其中和脂肪分化相關的信號通路有13條,包括cAMP信號通路、過氧化物酶體增殖物激活型受體(peroxisome proliferator activated-receptors,PPARs)信號通路、WNT信號通路、胰島素分泌、脂肪酸代謝、叉頭轉錄因子(the forkhead box O,FOXO)信號通路、脂肪酸生物合成、脂肪細胞脂解的調節、胰島素抵抗、MAPK信號通路、胰島素信號途徑、Janus激酶信號轉導與轉錄激活子(the Janus kinasesignal transducer and activator of tran-ions,JAK-STAT)信號通路、磷脂酰肌醇3激酶-蛋白激酶B(phosphatidylinositol 3'-OH kinase/protein kinase B,PI3K-AKT)信號通路。根據Q value從小到大排序(圖5b),前20富集的通路有:神經活性配體-受體相互作用、cAMP、鈣信號通路、細胞粘附分子(cell adhesion molecule,CAMs)、嗎啡成癮、膽汁分泌、胃酸分泌、安非他明成癮、蛋白質消化吸收、炎癥介質對TRP通道的調節、癌癥中的蛋白多糖、谷氨酸能突觸、醛固酮合成與分泌、胰腺分泌、可卡因成癮、癌癥中的microRNAs、醛固酮調節的鈉再吸收、心肌細胞腎上腺素能信號轉導、5-羥色胺能突觸、細胞因子-細胞因子-受體相互作用。

圖4 lncRNAs順式調控靶基因的預測Fig.4 Prediction of lncRNAs cis-regulated target genes
在共表達結果分析的基礎上,為了進一步獲得目的lncRNAs及其靶向基因,我們將DElncRNAs和DEGs之間符合PCC>0.7或PCC<-0.7且P<0.05的兩者定義為具有關系,并使用Cytoscape軟件構建了網絡互作圖。根據研究結果(圖6),我們共篩選出了31個差異lncRNAs,它們與134個DEGs相關,其中 1個 DElncRNA(TCONS_00027033)最多可靶向21個 DEGs,1個 DEG(ENSMUST00000114960)最多可與12個DElncRNAs共表達。

圖5 lncRNAs共表達靶基因的預測Fig.5 Prediction of lncRNAs co-expression target genes
2.7 qRT-PCR驗證結果
為了驗證測序數據的準確性,本研究使用qRTPCR對隨機選擇的3個lncRNAs和3個mRNAs進行定量驗證。測序結果中FC值>1或<1分別代表基因的上調或下調和表達量的升高或降低。結果表明qRT-PCR表達趨勢與測序結果一致,lncRNAs(ENSMUST00000152125、ENSMUST00000191042、TCONS_00033926)及 mRNAs(ENSMUST000000109-74、ENSMUST00000031327)在熒光定量PCR與測序結果中都上調,而mRNA(ENSMUST00000165806)下調(圖7)。這證明了本次測序結果的可靠性。

圖6 DElncRNAs與DEGs的共表達網絡互作圖Fig.6 Co-expression network mapping of DElncRNAs and DEGs

圖7 差異表達lncRNAs的驗證Fig.7 Validation of differentially expressed lncRNAs
3 討論
肥胖及其帶來的代謝類疾病已受到科學界的廣泛關注。目前應用于治療肥胖的方法包括飲食干預、運動治療、藥物治療及外科手術等[23],但上述療法具有一定的局限性和副作用[24]。近年來的研究表明,BAT激活增加能量消耗可作為防治肥胖癥的潛在靶點,通過激活BAT增加能量消耗是一種安全預防和控制肥胖的方法[25]。在模式動物中,增加和移植BAT能促進脂肪代謝,減輕體重,并改善全身胰島素的敏感性[7,26]。我們前期的研究表明,APS可以促進C3H10T1/2細胞的成脂分化,并提高其產熱能力。本研究旨在闡明APS改變了C3H10T1/2分化中lncRNA的表達譜,為從lncRNAs的角度揭示APS促進BAT細胞分化的分子機制提供科學依據。
RNA-seq技術的廣泛使用改變了我們對真核細胞轉錄組程度和復雜性的觀點。與其他方法相比,RNA-seq提供了更精確的轉錄物水平和轉錄物之間的聯系[27],已被廣泛地應用于 lncRNAs、micoRNAs和mRNAs等的研究中。lncRNAs的平均表達量、長度、外顯子個數均要小于蛋白編碼基因[28],這與我們的測序結果一致,進一步說明了本次測序結果的準確性。目前在lncRNAs順式靶基因預測中,選用的標準是lncRNAs上下游300 kb之內的基因定義為相關,但多選擇100 kb[29-30]。本次試驗我們采用了更加嚴苛的要求,篩選了上下游10 kb之內的基因定義為相關,這增大了獲得與BAT分化相關靶基因的概率。DElncRNAs的順式及共表達預測到的靶基因GO分析中,我們發現順式及共表達靶基因在分子功能本體中富集的條目保持著一致,這些條目包括核酸結合轉錄因子活性、轉錄因子活性、蛋白質結合等。這進一步證明了lncRNAs在基因組中具有調節基因表達的作用。
在對預測到的DElncRNAs順式調控靶基因進行KEGG分析后發現,靶基因在胰高血糖素信號途徑中富集顯著。胰高血糖素是一種主要來源于胰島α細胞的激素,在生理上通過獨特的受體來維持葡萄糖的穩態。研究發現,在BAT組織的切片中直接施用胰高血糖素會增加氧氣消耗和游離脂肪酸的釋放[31]。同時,胰高血糖素對BAT細胞的刺激可以誘導熱量的產生[32]。低溫環境下發現,敲除胰高血糖素基因的小鼠其BAT中的生熱標記(例如:Ucp1、Dio2和Ppargc1α)表達也在降低[33]。上述結果都證明了胰高血糖素對棕色脂肪細胞的代謝和分化具有重要的作用。外源性的APS可能通過改變相關lncRNAs的表達,調控多個處于胰高血糖素信號途徑的基因,從而影響BAT細胞的分化。在富集顯著程度居于前20的通路中,我們還發現胰島素分泌[34]、MAPK信號通路[35]、TNF等信號通路[36]都對BAT分化具有重要作用。在本研究結果中,前兩者信號通路中分別各有8個預測到的靶基因富集,后者有5個基因富集。綜上可知,DElncRNAs順式調控的靶基因富集于多條關于BAT分化的信號通路上的結果揭示了APS可能通過改變lncRNAs的表達來調控BAT的分化。
共表達靶基因KEGG通路分析結果表明,多個基因顯著地富集在了cAMP信號通路上。cAMP信號通路是一條調節脂肪細胞分化的經典途徑,體內的多種激素和細胞因子都是通過此途徑來調節脂質代謝和脂肪細胞分化的。細胞中cAMP的含量能決定蛋白激酶 A(protein kinase A,PKA)的活性,其能通過磷酸化2種脂質代謝重要的酶蛋白周脂素(perilipin A)和激素敏感脂酶(hormone-sensitive triglyceride lipase,HSL)來抑制脂肪細胞的分化[37]。同時細胞中cAMP濃度升高也可使磷酸二脂酶(phosphodiesterase,PDE)磷酸化從而被激活。PDE有14個亞基,分屬于7個家族[38],其中脂肪細胞中的PDE3B的活性與脂肪細胞分化呈正相關[39]。本研究還發現了多個靶基因富集在脂肪代謝的途徑,包括14個脂肪酸代謝,6個脂肪酸生物合成、6個不飽和脂肪酸的生物合成和6個脂肪酸降解,同時發現靶基因富集在了 PPAR信號通路、WNT信號通路[40]、JAKSTAT信號通路、PI3K-AKT信號通路等與脂肪分化的相關的通路中。這些結果進一步表明,添加APS導致的lncRNA表達譜變化對于BAT分化具有重要影響,且這種影響涉及到多個方面,并突出地顯示了APS可能主要通過影響cAMP信號通路來加速BAT細胞的分化。
在共表達網絡互作圖結果的基礎上,我們嘗試采用FDR<0.05且|log2FC|>1作為篩選條件進一步縮小差異lncRNAs的范圍來獲取目的lncRNAs。篩選共得到了10個DElncRNAs。在這10個lncRNAs中,我們聚焦了1個顯著下調的lncRNA(ENSMUST00000199606),其共表達分析預測靶向基因為ENSMUST00000209061,在對照組和處理組中表達呈顯著差異。ENSMUST00000209061基因編碼的是具有CCCH型RNA結合結構域且折疊成特殊的指狀結構的 RNA結合蛋白鋅指蛋白 36(tristetraprolin,ZFP36),其功能主要是通過與富含腺嘌呤和尿苷的mRNA 3'端結合來降解mRNA[41]。許多研究表明,ZFP36與脂肪分化及肥胖形成有關,并將其作為治療代謝疾病和肥胖的潛在靶點。在3T3-L1前脂肪細胞中,ZFP36能通過與絲裂原活化蛋白激酶磷酸酶 -1(mitogen-activated protein kinase phosphatase-1,MKP-1)mRNA結合來限制其表達,從而抑制脂肪細胞的分化[42]。與正常肥胖者相比,患有代謝綜合癥的肥胖者內臟脂肪中的ZFP36的蛋白和mRNA水平降低了4~5倍,且ZFP36基因位于19q13.1長臂,是個與代謝綜合征相關的區域[43]。有研究在人體中也發現ZFP36基因在網膜脂肪組織中的表達能預防胰島素抵抗和糖尿病[43]。更有趣的是,ZFP36包含許多絲氨酸和蘇氨酸,從而具備廣泛磷酸化的潛力,然而磷酸酶處理僅部分改變了蛋白質的遷移率,表明存在其他蛋白質修飾或與其他物質的結合阻止了磷酸酶功能[44]。而lncRNAs很重要的功能就是對于蛋白質的修飾,故我們猜想是lncRNA(ENSMUST0-0000199606)修飾了ZFP36,從而在脂肪分化和肥胖中起到重要作用。然而其真實的作用及具體的作用機制還需要進一步研究證明。
綜上所述,本研究結果揭示了APS能影響C3H-10T1/2細胞棕色脂肪化過程中lncRNAs的表達。靶基因預測顯示,APS可能主要通過DElncRNAs來影響包括胰高血糖素、cAMP等信號通路中的基因相對表達來促進BAT細胞分化,同時在進一步篩選中我們發現了差異lncRNA與ZFP36的關系。這些結果為進一步研究APS對BAT細胞分化的調控作用提供了科學依據。
猜你喜歡
鴨綠江(2021年35期)2021-04-19 12:24:18
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
中國生殖健康(2019年3期)2019-02-01 06:12:26
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
海軍航空大學學報(2015年3期)2015-11-11 17:20:00
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
終身教育研究(2014年5期)2014-02-28 01:23:06
