硫酸亞鐵法測定食鹽中亞鐵氰化鉀含量的不確定度評定
2020-11-23 01:48:28黎穎欣趙金利葉春常林少揚陳韻
食品安全導刊·下旬刊 2020年9期
黎穎欣 趙金利 葉春常 林少揚 陳韻
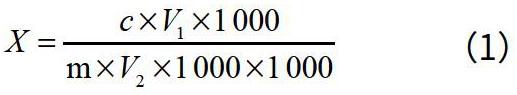

摘 要:對硫酸亞鐵法測定食鹽中亞鐵氰化鉀含量的過程進行不確定度評估,分析不確定度的主要來源,提出減小不確定度的建議。主要依據CNAS-GL006:2019《化學分析中不確定度的評估指南》,建立不確定度評估數學模型,對檢測過程引入的不確定度來源進行識別和定量。結果表明所選食鹽樣品中亞鐵氰化鉀含量為0.004 9 g/kg,擴展不確定度為0.000 5 g/kg,亞鐵氰化鉀的含量可表示為(0.004 9±0.000 5)g/kg(k=2)。經分析,對總不確定度貢獻較大的,包括標準溶液、回收率、重復性等引入的不確定度,可采取相應措施減小不確定度。
關鍵詞:硫酸亞鐵法;食鹽;亞鐵氰化鉀;不確定度評定
精制鹽和部分粉碎洗滌精制鹽的顆粒度較細且不均勻,而鹽本身又具有吸濕性,隨著存儲時間的延長,逐漸由松散的顆粒結成硬塊,對儲存、運輸和使用造成不便,因此需要添加適量的抗結劑。亞鐵氰化鉀又稱黃血鹽、黃血鹽鉀,由于價格低、所需用量少、抗結效果好,成為國內食鹽生產企業的首選抗結劑[1-2]。依據GB 2760-2014《食品安全國家標準 食品添加劑使用標準》[3]規定,亞鐵氰化鉀在鹽及代鹽制品的最大使用量為0.01 g/kg(以亞鐵氰根計)。實驗室對亞鐵氰化鉀的檢測結果,往往用作判定是否符合國標要求的依據。檢測結果準確度的重要性不言而喻。測量不確定度是評估檢測結果準確度和可信度的有效方法。
本研究按照GB 5009.42-2016《食品安全國家標準 食鹽指標的測定》[4]中的硫酸亞鐵法測定食鹽中的亞鐵氰化鉀,并根據CNAS-CL01-G003:2019《測量不確定度的要求》[5]、CNAS-GL006:2019《化學分析中不確定度的評估指南》[6]、JJF 1059.1-2012《測量不確定度評定與表示》[7]和JJF 1135-2005《化學分析測量不確定度評定》[8]的相關要求和方法,分析檢測過程的不確定度來源并分別量化,用以計算測量不確定度。此外,通過分析不確定度的主要來源,實驗室可采取相應的措施,減小測量不確定度,提高結果的可信度和可靠性[9-11]。
1 材料與方法
1.1 主要材料與試劑
食鹽:市售,加碘精制鹽;亞鐵氰化鉀(K4[Fe(CN)6]·3H2O)、硫酸亞鐵(FeSO4·7H2O)、硫酸:分析純,廣州化學試劑廠。
1.2 主要儀器與設備
MS304TS電子天平:梅特勒-托利多儀器(上海)有限公司;BSA2202S電子天平:賽多利斯(上海)貿易有限公司;7230G可見分光光度計:上海精密科學有限公司。
1.3 試驗方法
1.3.1 標準溶液和工作液的配制
準確稱取0.199 3 g亞鐵氰化鉀,溶于少量水,轉移入100 mL容量瓶中,加水稀釋至刻度,得1.0 mg/mL亞鐵氰根[Fe(CN)6]4-標準溶液。吸取上述標準溶液10.0 mL置于100 mL容量瓶中,加水稀釋至刻度,得0.10 mg/mL亞鐵氰根標準工作液。
1.3.2 標準工作曲線的繪制
分別吸取亞鐵氰根標準工作液0、0.1、0.2、0.3 mL、0.4 mL與0.5 mL,相當于0、10.0、20.0、30.0、40.0 μg與50.0 μg亞鐵氰根,分別置于25 mL比色管中,各加水至25 mL。以亞鐵氰根質量為橫坐標,對應的吸光度為縱坐標,繪制標準曲線。
1.3.3 試樣的制備和測定
稱取10.00 g食鹽樣品溶于水后轉移至50 mL容量瓶中,加水至刻度,混勻后過濾,棄去初濾液,然后吸取25.0 mL濾液于比色管中。試樣管與標準管分別加2 mL硫酸亞鐵溶液,混勻后放置20 min。用3 cm比色杯,以零管調節零點,于波長670 nm處測吸光值。根據試樣的吸光度,根據工作標準曲線計算測定用樣液中亞鐵氰根的含量。
1.3.4 回收率
準確稱取10.00 g食鹽樣品10份,在相同的實驗條件下進行制備和測定,計算回收率。
1.3.5 數學模型的建立
試樣中亞鐵氰化鉀的含量可按式(1)計算。
(1)
式(1)中:X為試樣中亞鐵氰化鉀(以[Fe(CN)6]4-計)的含量,g/kg;c為測定用樣液中亞鐵氰根的質量,μg;m為樣品質量,g;V1為樣品制備過程的定容體積;V2為用于試驗的濾液體積。
2 結果與分析
2.1 不確定度來源的識別和分析
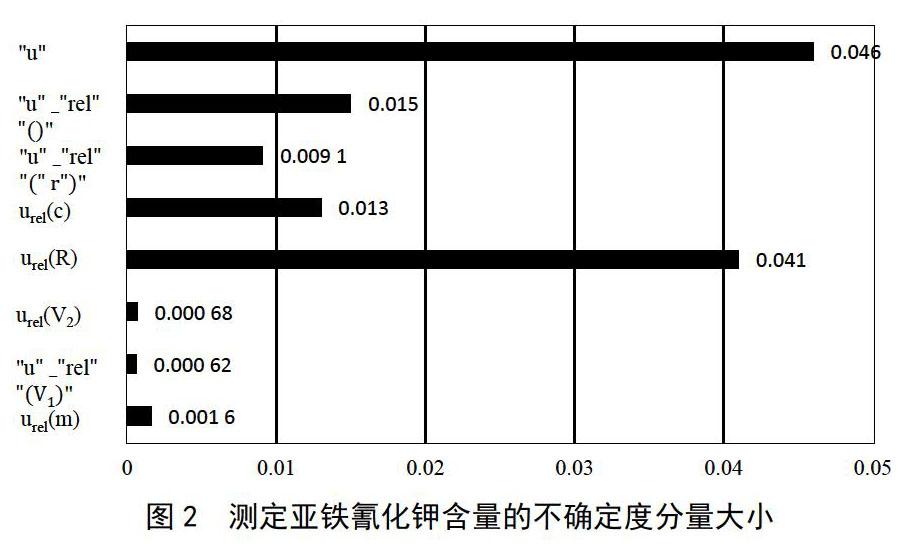
為了更好地識別所有的不確定度來源,本研究通過繪制因果關系圖,以亞鐵氰化鉀含量計算公式中的參數作為主要分支,再細致分析各參數的不確定度來源,將更多影響因素添加到此圖上。重復性評估作為一個整體,是對檢測全過程的評估,因此不再分別考慮樣品質量m、定容體積V1、濾液體積V2等參數中的重復性分量。為考察實驗過程的偏倚,增加了回收率評估。與亞鐵氰化鉀含量測定相關的不確定度來源分析見圖1。
