多壁碳納米管凈化-氣相色譜法測定瓜菜中六六六和滴滴涕殘留研究
2020-12-07 08:46:56李解樊磊
現代農業科技 2020年19期
關鍵詞:固相萃取
李解 樊磊
摘要 ? ?本文建立了超聲波提取、固相萃取凈化、氣相色譜-電子捕獲檢測器法(GC-ECD)同時測定瓜菜中α-六六六、β-六六六、γ-六六六、δ-六六六、p.p′-DDE、p.p′-DDD、p.p′-DDT和o.p′-DDT等8種有機氯農藥殘留的方法。樣品用丙酮+正己烷(體積比1∶1)混合溶液超聲提取,采用自制多壁碳納米管/弗羅里硅土固相萃取柱凈化,GC-ECD測定,外標法定量。結果表明,8種有機氯農藥在0.5~500.0 μg/L濃度范圍內線性關系良好,相關系數r均大于0.999;添加水平在1.25~100.00 μg/kg時,回收率在81.5%~113.2%之間,相對標準偏差(n=5)在2.29%~14.48%之間,檢出限為0.1~0.6 μg/kg,定量限為1.25~5.00 μg/kg。該方法操作簡單,前處理凈化效果好,具有較好的準確度、精密度和靈敏度,適用于同時測定瓜菜中8種有機氯農藥。
關鍵詞 ? ?多壁碳納米管;固相萃取;氣相色譜法;滴滴涕;六六六;瓜菜
中圖分類號 ? ?S481+.8 ? ? ? ?文獻標識碼 ? ?B
文章編號 ? 1007-5739(2020)19-0110-04
六六六和滴滴涕屬有機氯類農藥,曾作為高效廣譜殺蟲劑在全球范圍內廣泛使用。但此類化合物毒性強,具有“三致”毒性以及免疫、神經、內分泌干擾等多種毒性效應[1],雖然20世紀70年代后期在全世界范圍內禁用,由于很難降解,至今在土壤[2]、水體[3]、大氣[4]、食物樣品[5]等多種介質中仍頻繁檢出,部分地區殘留量仍然較高,嚴重威脅人類健康。因此,開展此類農藥殘留的檢測研究意義重大。
在六六六和滴滴涕農藥殘留分析方法中,樣品的前處理是其檢測的關鍵,常用的前處理技術有索氏提取[6]、QuEChERS[7]、加速溶劑萃取[8]、固相萃取[5-6]、固相微萃取[9]等,其中,固相萃取是目前應用較多的前處理技術之一。由于固相萃取的效果顯著受萃取填料性質的影響,因而發現和應用新型萃取材料是該技術研究重點之一。碳納米管是一種新型納米碳材料,具有獨特的中空結構和大比表面積等特征[10],近年來在農產品質量安全檢測領域廣泛應用[11],并獲得了優異的效果,但在瓜類蔬菜中滴滴涕和六六六殘留檢測上的研究報道較少。本文以黃瓜為瓜菜樣品代表,采用多壁碳納米管作為固相萃取主要填料,與氣相色譜-電子捕獲檢測器法(GC-ECD)聯用,建立了同時準確測定4種六六六(α-六六六、β-六六六、γ-六六六、δ-六六六)和4種滴滴涕(p.p′-DDE、p.p′-DDD、p.p′-DDT、o.p′-DDT)殘留量的方法。該方法各項技術指標均滿足檢測要求,為瓜菜中8種有機氯農藥的同時檢測提供了參考方法。
1 ? ?材料與方法
1.1 ? ?儀器與試劑
主要試驗儀器:7890A氣相色譜儀,配電子捕獲檢測器(ECD)及Chemstation工作站(美國Agilent公司),KQ-500DE超聲波清洗器(昆山超聲儀器公司),HSC-24B氮吹儀(天津恒奧科技公司)。
主要試劑:農藥標準溶液α-六六六、β-六六六、γ-六六六、δ-六六六、p.p′-DDE、p.p′-DDD、p.p′-DDT、o.p′-DDT,質量濃度均為100 mg/L,購于農業農村部環境保護科研監測所。多壁碳納米管(長度為10~30 μm,外徑為10~20 nm)(南京先豐納米材料科技有限公司);弗羅里硅土(農殘級)(美國ROE公司);正己烷(色譜純,瑞典歐普森);氯化鈉、丙酮(分析純,上海國藥集團);無水MgSO4(分析純),使用前于620 ℃下灼燒4 h。試驗用水為去離子水。
1.2 ? ?試驗方法
1.2.1 ? ?標準溶液配制。根據8種待測農藥在ECD檢測器上的響應值,取p.p′-DDE、β-六六六單標溶液各0.5 mL,o.p′-DDT、p.p′-DDT單標溶液各1.0 mL,其余農藥單標溶液各0.25 mL于10 mL容量瓶中,用正己烷定容,得混合標準儲備液,放置于4 ℃冰箱待用。使用時,分別用正己烷、空白基質提取液逐級稀釋,配制不同質量濃度的溶劑和基質匹配標準工作溶液。
1.2.2 ? ?固相萃取小柱制備。弗羅里硅土填料經620 ℃烘燒4 h后冷卻備用,使用前經130 ℃烘燒2 h,然后加入5%水去活化。采用干法裝柱,取6 mL固相萃取小柱,由下至上依次裝入篩板、1 g弗羅里硅土和0.03 g多壁碳納米管填料、篩板,壓實,小柱待用。
1.2.3 ? ?樣品前處理。樣品按四分法取樣,勻漿后備用。
提取:準確稱取試樣3.0 g至50 mL塑料離心管中,加入7.5 mL丙酮+正己烷(體積比1∶1)溶液和1 g氯化鈉,渦旋2 min,超聲波法提取15 min,加入2 g無水MgSO4,渦旋2 min,于8 000 r/min下離心4 min,移取上清液于另一潔凈的離心管中,樣品殘渣按上述操作重復提取1次,合并2次提取液,準確移取5 mL氮吹濃縮至約1 mL,待凈化。
凈化:將固相萃取小柱用10 mL丙酮+正己烷(體積比1∶9)溶液預淋洗后,棄去流出液,加入待凈化樣品溶液,重力過柱。用10 mL丙酮+正己烷(體積比為1∶9)溶液洗滌溶解樣品的離心管,并淋洗小柱,收集全部流出液,在70 ℃水浴下氮吹至近干,用正己烷定容至1 mL,混勻,過0.22 μm濾膜后,供GC-ECD測定。
1.2.4 ? ?氣相色譜條件。色譜柱為DB-1701(30.00 m×0.32 mm×0.25 μm);進樣口溫度為250 ℃;升溫程序為150 ℃保持0.5 min,然后以8 ℃/min升溫至240 ℃,再以3 ℃/min升至260 ℃保持2 min;ECD檢測器溫度為300 ℃;載氣為高純氮氣,流速為1.7 mL/min;進樣量為1.0 μL,不分流進樣。
2 ? ?結果與分析
2.1 ? ?色譜條件選擇
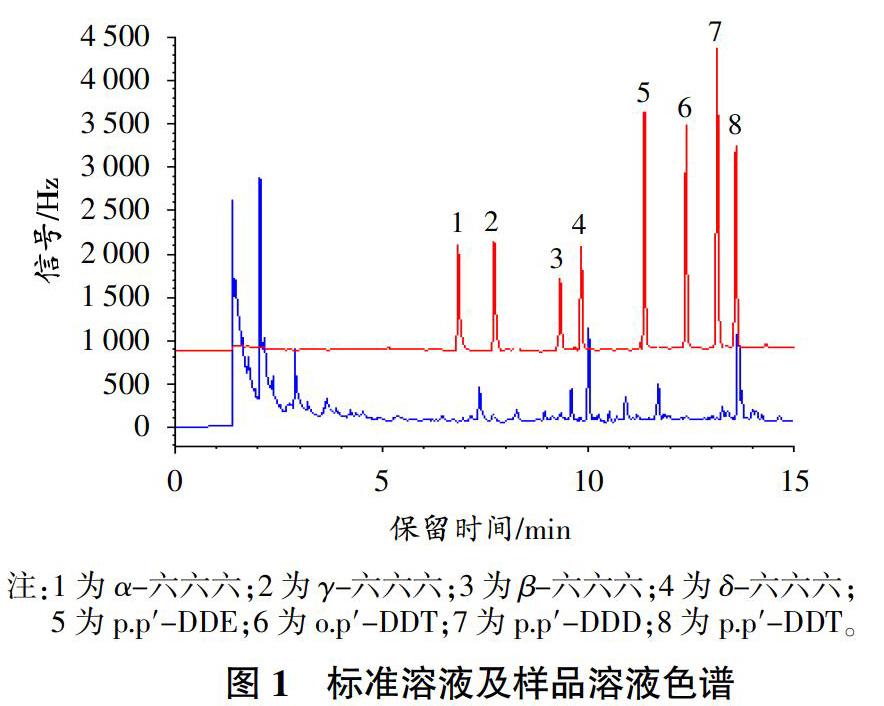
根據待測農藥的性質,通過改變色譜柱種類(DB-1701、HP-5)、進樣口溫度(220、230、240、250 ℃)、載氣流速(1.0、1.2、1.5、1.7 mL/ min)、升溫程序等條件,觀察各農藥的信號強度和分離分析情況,最終選擇1.2.4氣相色譜條件。在此色譜條件下,標準溶液和實際樣品溶液色譜圖如圖1所示。可以看出,8種農藥在15 min內能夠完全分離且峰型較好,組分出峰處沒有明顯的干擾雜峰,表明色譜條件選擇合適。
2.2 ? ?前處理條件選擇
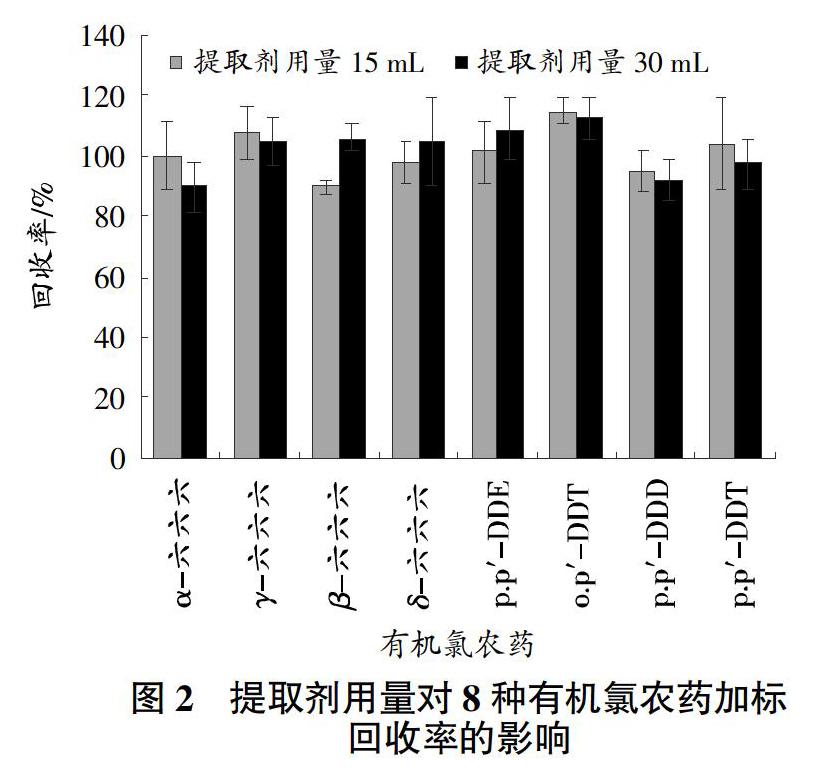
超聲法操作簡單,可實現多個樣品同步處理,故選擇該方式提取樣品。有機氯農藥屬于弱極性化合物,適合用非極性的正己烷為提取劑,同時加入適量的丙酮,可增強對樣品的滲透性,提高萃取效率。比較了丙酮+正己烷(體積比1∶1)、丙酮+正己烷(體積比1∶2)作為提取劑對8種有機氯農藥加標回收率的影響。結果顯示,采用上述2種提取劑時,8種農藥的回收率分別為85.6%~110.2%、76.3%~95.5%,故選擇丙酮+正己烷(體積比1∶1)為提取劑;進一步試驗了其用量分別為15 mL和30 mL時對待測農藥加標回收率的影響(圖2),發現兩者的回收率相差不大,分別為89.8%~114.7%、89.7%~112.7%,均滿足農藥殘留檢測的要求[12],為減少溶劑的消耗,選擇提取劑用量為15 mL。
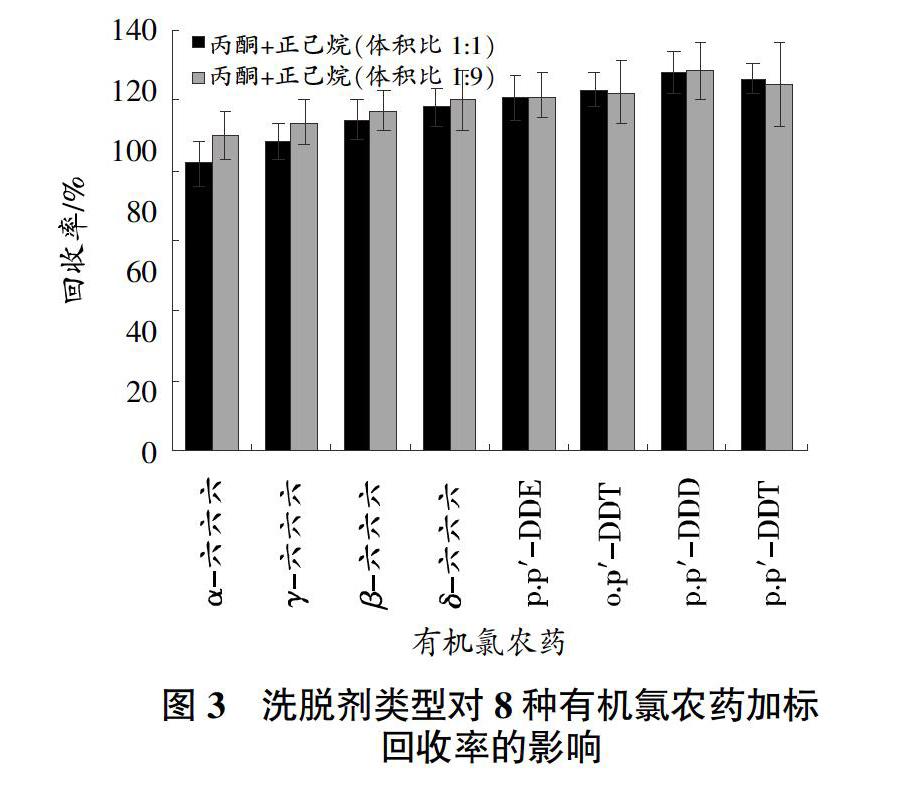
采用固相萃取小柱凈化樣品,比較了商品化的弗羅里硅土柱和自制的多壁碳納米管/弗羅里硅土柱對樣品提取液的凈化效果。試驗發現,提取液通過商品化弗羅里硅土柱凈化,洗脫液呈淡黃色,存在色素干擾;通過自制多壁碳納米管/弗羅里硅土柱凈化,色素類物質被多壁碳納米管吸附,洗脫液澄清透明,有效減少了色素干擾,故選擇自制的多壁碳納米管/弗羅里硅土柱進行固相萃取凈化。比較10 mL體積比分別為1∶1和1∶9的丙酮+正己烷混合溶液作為洗脫劑對8種有機氯農藥洗脫效果的影響。從圖3可以看出,采用體積比分別為1∶1和1∶9的丙酮+正己烷混合溶液洗脫劑洗脫,8種有機氯農藥的回收率分別為81.9%~108.0%、89.9%~108.7%,后者總體優于前者,故選擇10 mL丙酮+正己烷(體積比1∶9)混合溶液作為洗脫劑。
2.3 ? ? 基質效應分析
取1.2.1中不同質量濃度的溶劑和基質匹配標準工作溶液進行測定,通過比較2條標準曲線的斜率判斷基質效應(ME)的強弱。當|ME|<20%時,為弱基質效應,無須采取補償措施;當20%≤|ME|≤50%時,為中等程度基質效應,需采取措施補償基質效應;當|ME|>50%時,為強基質效應,需采取措施補償基質效應[13]。計算公式如下:
ME(%)=(基質匹配標準曲線斜率/溶劑標準曲線斜率-1)×100
從表1可以看出,8種待測農藥的|ME|值均在20%以內,為弱基質效應。因此,本方法采用純溶劑配制標準溶液進行定量分析。
2.4 ? ?方法線性范圍、檢出限及定量限
按本方法色譜條件,對8種待測農藥的溶劑標準工作溶液進行測定,以各農藥組分的峰面積(y)對質量濃度(x)繪制標準曲線,線性范圍、回歸方程和相關系數見表1。結果表明,8種農藥在各自濃度范圍內線性良好,相關系數在0.999 1~0.999 9之間。對最低添加水平的樣品進行前處理和測定,以色譜圖中信噪比的3倍確定各農藥的檢出限,以最小添加濃度確定其定量限(表1)。
2.5 ? ?方法的回收率及精密度
以空白黃瓜為代表基質,在高、中、低3個水平下進行添加回收試驗,每個添加水平做5次平行,分別測得回收率及相對標準偏差。8種待測農藥的平均回收率為81.5%~113.2%,相對標準偏差為2.29%~14.48%(表2), 表明方法可靠、穩定。
2.6 ? ?樣品測定
從當地超市購買黃瓜、西葫蘆、絲瓜等瓜菜樣品,按本試驗方法進行測定,未檢出待測農藥殘留。同時分別添加中等水平的8種農藥標準溶液進行添加回收試驗,結果見表3。8種農藥的添加回收率在85.9%~116.6%之間,相對標準偏差在3.05%~15.80%之間,符合農藥殘留分析的要求[12],說明該方法具有一定的普及性。
3 ? ?結論
本文以新型納米材料多壁碳納米管和弗羅里硅土為填料,通過自制固相萃取小柱,與GC-ECD法聯用測定瓜菜中8種典型有機氯農藥的殘留量,獲得了較好的分析結果。在優化的檢測條件下,方法的回收率為81.5%~113.2%,相對標準偏差(n=5)小于14.5%,檢測限為0.1~0.6 μg/kg,定量限為1.25~5.00 μg/kg,具有較好的準確度、精密度和靈敏度,適用于瓜菜中8種有機氯農藥同時準確測定。
4 ? ?參考文獻
[1] 曾鴻鵠,覃如瓊,莫凌云,等.有機氯農藥對人體健康毒性研究進展[J].桂林理工大學學報,2014(3):549-553.
[2] 邵波,劉勇,李鑫,等.長三角地區土壤中有機氯農藥殘留量及其分布特征[J].環境化學,2018,37(4):204-215.
[3] 陳衛平,彭程偉,楊陽,等.北京市地下水有機氯和有機磷農藥健康風險評價[J].環境科學,2018,39(1):120-125.
[4] 彭成輝.寧波市東部地區大氣中有機氯污染物的時空分布和污染特征[J].農業環境科學學報,2019,38(4):787-797.
[5] 孟媛,劉翠翠,仇雁翎,等.上海市稻米中有機氯農藥殘留水平及健康風險評價[J].環境科學,2018,39(2):927-934.
[6] 呂愛娟,時磊,沈小明,等.索氏提取-全二維氣相色譜法測定化工區江灘沉積物中20種有機氯農藥和7種多氯聯苯的含量[J].理化檢驗(化學分冊),2020,56(1):20-25.
[7] 李福敏,邵林.QuEChERS-氣相色譜-三重四級桿串聯質譜法測定茶葉中的12種有機氯農藥殘留[J].食品安全質量檢測學報,2018,9(6):169-174.
[8] 趙君,吳坤,蘭世超.加速溶劑萃取-氣相色譜法測定刺梨中11種有機氯農藥[J].廣州化工,2019(13):120-122.
[9] 虞游毅,楊璐,廖享,等.固相微萃取-氣相色譜質譜聯用法測定蘋果中4種有機氯類農藥殘留[J].農藥,2018,57(1):54-57.
[10] 趙昌平,楊葉琴,朱強,等.碳納米管在食品農藥多殘留測定中的應用[J].食品安全質量檢測學報,2019,10(13):4382-4387.
[11] 劉騰飛,楊代鳳,毛健,等.碳納米管材料在食品安全分析中的應用[J].化工進展,2018,37(10):3699-3725.
[12] 中華人民共和國農業農村部.農作物中農藥殘留試驗準則:NY/T 788—2018[S].北京:中國農業出版社,2018.
[13] 李瑩,韓梅,邱世婷,等.QuEChERS/超高效液相色譜-串聯質譜法測定土壤中31種磺酰脲類除草劑殘留[J].分析測試學報,2020,39(3):343-350.
猜你喜歡
分析化學(2016年7期)2016-12-08 00:54:07
中國科技博覽(2016年2期)2016-04-25 14:11:43
湖北工業職業技術學院學報(2016年1期)2016-04-20 17:12:54
分析化學(2015年10期)2015-11-03 07:52:24
食品安全導刊(2015年10期)2015-10-26 04:44:22
安徽農學通報(2015年18期)2015-10-20 00:50:11
安徽農學通報(2015年17期)2015-09-30 00:52:24
分析化學(2015年9期)2015-09-11 07:09:54
肉類研究(2015年5期)2015-08-08 12:46:08
肉類研究(2015年3期)2015-06-16 12:40:36
