醫療器械執行標準協調性探討
2020-12-22 03:51:04李寶林
中國醫藥科學 2020年22期
李寶林
河北省藥品醫療器械檢驗研究院,河北石家莊 050200
我國醫療器械注冊管理經歷了一系列變革,在相關規章文件以及執行標準的應用實踐中,呈現出了一些不協調的現象。探討當前醫療器械標準及“產品技術要求”的有關規定和運行狀況,尋求改進之處。
1 標準化法律法規——國標、行標
1.1 標準化管理
我國標準化體系包括國家標準、行業標準以及地方標準、團體標準、企業標準,由國家標準化管理委員會統一管理。
我國標準化法(2017年修訂版[1])將國家標準分為強制性標準和推薦性標準,其修訂之一即原標準化法(1988年版[2])將國家標準、行業標準區分為強制性標準和推薦性標準,而新版本則將行業標準、地方標準歸均屬于推薦性標準。目前全國標準信息公共服務平臺(http://std.samr.gov.cn/)共收錄67種行業標準,如醫藥(YY)、中醫藥(ZY)、衛生(WS)。
各類產品標準通常由技術要求、檢驗方法、檢驗規則等技術要素及標準編號、標準名稱、適用范圍、規范性引用文件、術語和定義、標志、包裝、運輸與貯存、附錄等要素組成。
1.2 醫療器械標準
醫療器械標準沒有單獨的標準體系,早先按照國家標準化體系管理。2000年首次發布實施的《醫療器械監督管理條例》[3]規定醫療器械應當執行醫療器械國家標準,對于尚無醫療器械國家標準的產品應當符合醫療器械行業標準;同時指明,醫療器械國家標準由我國標準化主管部門會同藥品監督管理部門制定,醫療器械行業標準由藥品監督管理部門制定。
《醫療器械監督管理條例》修訂版(國務院令650號[4])明確了醫療器械產品應當執行強制性醫療器械國家標準,若沒有強制性國家標準,則執行醫療器械強制性行業標準。
修訂版條例不再強調行業標準的制定部門,不言而喻,即按照我國已建立的國家標準化體系進行管理。我國標準化法規定需要在全國行業范圍內統一、且尚無推薦性國家標準可由有關行政主管部門制定行業標準,并由標準化行政主管部門備案。
1.3 沿革
2001 年前,按照我國標準化管理要求,醫療器械執行國家、行業或企業標準。2002~2014年,醫療器械執行標準有國家、醫藥行業、注冊產品標準。2015年后,以“醫療器械產品技術要求”為執行依據。
2 醫療器械行政規章——執行標準的管理
2.1 部門規章
原國家食品藥品監督管理總局修訂版《醫療器械標準管理辦法》(令第33號[5])定義醫療器械標準為“由國家食品藥品監督管理總局依據職責組織制修訂,依法定程序發布,在醫療器械研制、生產、經營、使用、監督管理等活動中遵循的統一的技術要求”。此定義中的“技術要求”一詞系指標準要素中產品安全、功能等性能參數或管理要求。
該辦法將醫療器械標準劃分為強制性標準、推薦性標準。提出了制定強制性、推薦性標準的范圍,強調有關研發、生產、銷售、使用醫療器械應嚴格執行強制性醫療器械標準。
醫療器械行業標準制修訂工作由原國家食品藥品監督管理總局組織、發布、監管,由醫療器械標準管理中心履行具體職責,并成立相應的標準化技術委員會負責技術工作。
2.2 醫療器械產品技術要求
國務院650號令規定了醫療器械產品備案或產品注冊須提交“產品技術要求”。取消了此前執行的“醫療器械注冊產品標準”,即原國家食品藥品監督管理局16號令[6]指出的經由受理注冊的國家、省、市級藥監部門復核的產品標準。
原國家食品藥品監督管理總局發布了第4號令[7],指出醫療器械應滿足經產品注冊核準或產品備案的“產品技術要求”。此處的“產品技術要求”一詞屬于專用名詞,首次成為醫療器械產品的執行標準,實施行政許可。
第4號令提出“產品技術要求”不得低于適用的強制性國家標準、行業標準。“產品技術要求”由醫療器械注冊人編制,經受理注冊的國家、省級藥監部門于批準注冊時對第三類、第二類醫療器械“產品技術要求”予以核準;對第一類醫療器械實施備案,未提出核準要求。
2.3 產品技術要求的要素
申報注冊的“產品技術要求”文件名稱采用申請注冊/備案的產品名稱,編號采用相應的產品注冊證號/備案號,其內容由四部分組成,即產品規格型號及劃分說明、性能指標、檢驗方法、術語。性能指標系指醫療器械的功能性、安全性等質量控制要求,須首先選擇國家、行業標準的方法。要求(第三類)體外診斷試劑在其技術要求中以附錄的形式標明主原料、中間成品、生產加工工藝的技術要求。其編制、評價、核準、應用、修訂/變更等要求,見表1。
3 實施要求討論——標準體系與作用
3.1 醫療器械行業標準屬性
第33號令明確了醫療器械行業標準代號的要求:“由大寫漢語拼音字母構成”“強制性行業標準的代號為‘YY’,推薦性行業標準的代號為‘YY/T’”。此辦法中的“YY”與“醫療器械”或“醫療器械行業標準”的漢語拼音不甚相關。而此“YY”代號與標準化管理體系已有的醫藥行業標準的代號“YY”(“醫藥”二字的漢語拼音字頭)相重,無法區別,致使標準代號失去了唯一性;或者說該辦法中定義的“醫療器械行業標準”是個虛擬的標準體系,而屬于“醫藥行業標準”范疇。若是,則與標準化工作重疊,但該辦法由原國家食品藥品監督管理總局發布,而非國家標準化管理部門發布,屬于管理不當所致。
有關醫療器械標準實例,例一,《一次性使用無菌牙科注射針》[8]標準封面信息:分類號ICS 11.040.20,C31;編號 YY/T 0587-2018,中華人民共和國醫藥行業標準,2018-04-11發布,2019-05-01實施,國家藥品監督管理局發布;由中國標準出版社發行,2018年6月第一版。前言:由國家藥品監督管理局提出,由全國醫用注射器(針)標準化技術委員會(SAC/TC95)歸口。例二,國家2019年第5號(總第233號)標準備案公告[9]顯示,《醫療器械唯一標識基本要求》YY/T 1630-2018,狀態為現行推薦性醫藥行業標準,中國標準分類號C30,國際標準分類號11.040.01;35.040,技術歸口國家藥品監督管理局醫療器械標準管理中心,批準發布部門為國家藥品監督管理局。
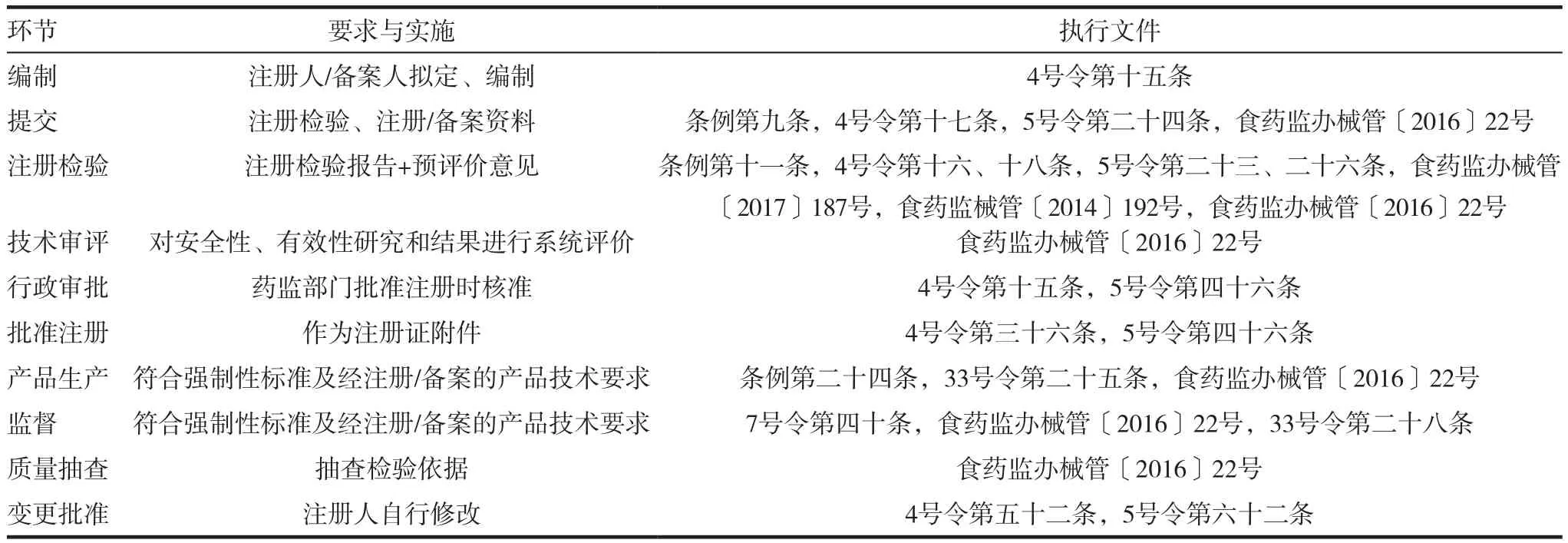
表1 編制、評價、核準、應用匯總表
這兩個標準,一個是具體的產品類醫療器械標準,一個是醫療器械通用要求的管理標準。確然可見,在實際實施過程中該法所稱的醫療器械行業標準即屬于我國標準管理體系中的醫藥行業系列標準(YY、YY/T)。
3.2 “產品技術要求”的作用以及監管
對產品注冊和生產的要求,修改版條例(國務院令680號[10])規定生產企業應有效地運行質量管理體系,應按照經產品注冊或經產品備案的醫療器械“產品技術要求”實施生產,并確保其符合“強制性標準以及經注冊或者備案的產品技術要求”。按照注冊管理辦法的要求,在實際工作中均強調以“產品技術要求”的形式提交申報產品注冊或者產品備案資料。
原國家食品藥品監督管理總局文件(食藥監辦械管〔2017〕187號[11])要求檢驗機構在注冊檢驗時,“嚴格按照”食藥監械管〔2014〕192號[12]文件的要求對醫療器械產品技術要求開展預評價工作,并要求“充分考慮國家標準、行業標準的完整性和適宜性”。強調依據“產品技術要求”進行產品注冊申請。而在實踐中將中華人民共和國藥典、國家標準、行業標準主要用于針對“產品技術要求”實施預評價活動,而未將已發布的適用醫療器械產品的“強制性”國家標準或行業標準作為控制醫療器械產品質量的技術依據。可見,在應用中強調“產品技術要求”,而消弱或未能使國家標準、行業標準發揮其在一定范圍內共同遵守和統一技術規范的作用,也失去了標準化的意義。
對日常生產監督的要求,原國家食品藥品監督管理總局發布第7號令[13]指出生產商應依據“經注冊或者備案的產品技術要求組織生產,保證出廠的醫療器械符合強制性標準以及經注冊或者備案的產品技術要求”。此后,原國家食品藥品監督管理總局發布工作文件(食藥監辦械管〔2016〕22號[14])進一步強調“嚴格按照……”;4號令第二十五條也提出同樣要求,而且第二十八條指示食品藥品監督管理部門對制造商執行“醫療器械強制性標準以及經注冊或者備案的產品技術要求”的情況進行監督檢查。要求按“產品技術要求”組織生產,在監督中強調“符合強制性標準”,令人茫然。
3.3 應用實況與管理不協調
在產品的研制階段要求管理體系滿足醫療器械生產質量管理規范并有效運行,經過注冊申請人驗證、確認其有效性和符合性,進而形成“產品技術要求”提交產品注冊,這是設計開發的輸出結果之一。在申請注冊過程中,經藥品監督管理部門組織相關人員對該管理體系的執行情況及研制的真實性實施了注冊核查,并出具核查報告。在注冊檢驗階段,由承擔注冊檢驗的機構按照預評價工作要求,針對性能指標的完整性、適用性;檢驗方法可操作性、重復性、適應性;所引用強制性國家標準或者行業標準的完整性、與申報產品的適宜性、所引條款的適用性;引用中華人民共和國藥典的完整性、適宜性、適用性等方面實施評價和檢驗活動,并要求同時出具注冊檢驗報告書、預評價意見。在技術審評階段,由技術審評機構組織評審人員或邀請相關專家對注冊申請資料給予全面審核,得出結論性審核意見,并提交技術評審報告。據此而論,經過上述3個階段形成的“產品技術要求”可謂對是否“符合強制性標準”作出了較為完整的技術審核。而仍然要附加“符合醫療器械強制性標準”并監督“醫療器械強制性標準”的執行情況,監督對象不甚明了、監管人員技術能力難以滿足,也似乎是對上述一系列驗證、試驗、評價、審評工作的作用和必要性的否定。
在技術審評報告和注冊核查報告的基礎上,再經藥品監督管理部門核準、審批,完成行政許可決定,將“經過核準的產品技術要求”作為附件隨醫療器械產品注冊證一同核發給申請人。即使對于第一類實施備案管理的醫療器械“產品技術要求”也是經藥品監督管理部門備案后生效的,均是行政許可的最終結果。無可否認,這一行政許可結果——“產品技術要求”即是組織醫療器械生產和控制產品質量唯一的、合法的技術依據。然則,反復謹慎地提出是否“符合醫療器械強制性標準以及…”,恰似對上述行政許可的質疑。要求按“經注冊或者備案的產品技術要求組織生產,保證出廠的醫療器械符合強制性標準以及……”混淆了“產品技術要求”與“醫療器械強制性標準”,模糊了制定“產品技術要求”的意義,也是對上述行政許可結論的否定。
核發的醫療器械產品注冊證書附件——“產品技術要求”存在無已核準標識的現象,或者有的僅加蓋“此文件由申請人或注冊人提供”條章的實例,注冊證書附件與其證明作用不協調。不利于在行政執法中有效應用,也突顯了將“產品技術要求”的管理責任浮于表面的現狀。假若欲將行政監管責任由企業管理、檢驗評價、技術審評、行政審批環節再分解到督查環節,則無責任可言,實為不可。
對于“產品技術要求”中引用的國家/行業標準修訂情況的監督,則屬于注冊變更,4號令、5號令[15]均劃歸于許可事項變更,卻提出由注冊人“根據變更內容自行修改產品技術要求”,行政許可事項與實際執行不協調。將產品注冊證附件——行政許可結果,委之于注冊人“自行修改……”的做法,即缺乏嚴肅性,又失之于行政許可的規范化。
“產品技術要求”在我國檢驗檢測機構資質認定以及國家實驗室認可申請中也不能得到受理。
4 結論與建議
有關醫療器械的標準采用翻譯法等同采用國際標準而轉化為醫藥行業標準者眾多。眾所周知,國際/國外標準與我國標準在格式、表述模式、要求等方面有些許的差異,表現在多數轉化標準為通用性基本要求,在我國可操作性差。提升已有醫療器械標準化技術委員會的能力,制定出適合于我國醫療器械行業發展的執行標準,尤其是結構、用途明確,工藝成熟的產品標準,提高標準的實用性,使之能有效應用于產品注冊和日常監管。
醫療器械標準未能列入我國藥品標準的管理系統,也未按照我國標準化體系管理。醫療器械產品的組成結構賦予了其具有安全性和有效性的性能特點,在強調特殊性的同時,不宜偏離已建立的國家標準化管理體系。鑒于醫療器械的特性和部門規章中標準管理要求的諸多不協調,建議控制創新探索步伐,堅持我國標準化法統一的管理體系,加強標準化工作的協同性,規范醫療器械標準的制定、應用與管理。執行標準是監管的技術依據,不可不慎。
在歷次機構改革中,藥監部門變動較大,也保持了專業領域的獨立性,建議保持管理規章的連貫性,促進醫療器械行業健康發展。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
口腔護理用品工業(2021年4期)2021-11-02 08:22:56
當代陜西(2019年8期)2019-05-09 02:22:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
中國公路(2017年9期)2017-07-25 13:26:38
專用汽車(2016年4期)2016-03-01 04:13:43
Coco薇(2015年1期)2015-08-13 02:23:50
汽車維修與保養(2015年8期)2015-04-17 03:32:51
中國質量與標準導報(2014年9期)2014-02-28 22:25:45
玩具(2009年10期)2009-11-04 02:33:14
