進化樹在細菌親緣關系分析中的應用研究
2021-01-04 03:28:12遲文靜劉宜昕張艷梅
檢驗醫學 2020年12期
關鍵詞:分析
遲文靜, 劉宜昕, 王 粟, 劉 濤, 趙 虎, 張艷梅
(復旦大學附屬華東醫院檢驗科,上海 200040)
分子系統發育學是利用分子特征研究生物體之間進化關系的學科,在揭示生物進化途徑、研究生物多樣性和分子流行病學特征、鑒定菌種和基因功能等方面發揮重要作用[1]。早期微生物分子系統發育研究依賴于蛋白質序列,其中應用廣泛的蛋白質序列有鐵氧還蛋白和細胞色素等。20世紀70年代中期,有學者開始使用16SrRNA的基因序列構建進化樹,以確定不同生物之間的進化關系。目前,隨著高通量測序(next generation sequencing,NGS)等技術的應用,大大降低了基因分析的成本,加快了基因分析的速度,為細菌進化樹的構建提供了更多類型的分析和展示形式[2]。本文對進化樹及其在細菌親緣關系中的應用進行綜述。
1 進化樹概述
1.1 進化樹定義
進化樹又稱系統發生樹,是描述生物體形成或進化順序的拓撲樹結構,通常是二叉樹的形狀,一般由一系列節點和分支組成,節點代表某個具體序列,節點之間的連線代表物種之間的親緣關系[3]。構建進化樹不僅需要分子生物學、遺傳學、生態學等生命科學學科知識,還需要統計學、計算機學等多個學科知識的融合[4]。微生物進化樹的構建過程主要有:序列數據的獲取、進化距離模型的確定、多個序列的比對、對比后結果的提取以及算法和參數的選擇[5]。進化樹可以分析未知細菌和已知細菌間的親緣關系[6],在遺傳本質上探究細菌多樣性的產生機制。
1.2 進化樹的呈現方式
根據是否制定根節點,進化樹可以分為有根樹和無根樹2種呈現方式[7]。有根樹制定了根節點,從樹中可以看出各節點之間的距離和各分支分化的先后關系。有根樹引入外群作為根節點,而外群通常選擇與研究序列關系密切的序列,且能很好地聚類;或者選擇比研究序列進化歷史更早的序列,故有根樹可以看到不同細菌間關系的遠近,還可以看到細菌的進化順序和方向[8]。無根樹不引入外群,沒有根節點,只能看出個各個節點的拓撲結構和相對距離,因而無根樹功能單一,可以顯示不同細菌的聚類關系和相對距離的遠近,卻無法顯示細菌的起源和進化方向[9]。見圖1。
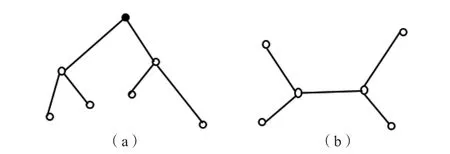
圖1 進化樹的呈現方式
1.3 進化模型
進化模型是對微生物進化變異進行的數學描述,描述內容主要有:基因的點突變、插入缺失,各堿基突變發生的概率及核苷酸組成頻率等和目標菌株間的親緣關系,選擇和構建合適的進化模型是研究細菌進化的前提[10]。生物進化的研究按層次可分為宏進化和微進化。宏進化是細菌間的進化,主要指細菌不同目/科/屬/種的進化[11],微進化是指細菌種內或近緣菌種間的進化[12]。以幽門螺桿菌為例,在其微進化過程中,構建幽門螺桿菌不同菌株間的進化樹模型,不僅需要估計堿基點突變率,還需要估計重組率、核苷酸差異率及重組核苷酸片段長度等重要信息[13]。對于細菌的進化分析,選擇合適的進化模型才可能得到可靠的結果,反之可能得到不準確甚至是錯誤的結果,這種現象主要是由進化樹不相同的拓撲結構或分支長度造成的,因此選擇合適的進化模型、構建正確的進化樹至關重要。
1.4 進化樹分析方法
1.4.1 距離矩陣法 距離矩陣法是一種以細菌核苷酸序列間的變異估計菌株間距離,并通過距離矩陣構建進化樹的方法。這種方法首先需要將輸入的核苷酸序列數據轉化為距離信息,然后通過距離信息進一步構建進化樹,主要分為系統樹法和網絡法[14]。總的來說,距離矩陣法是一種基于距離構建進化樹的方法,其優點在于簡單、直觀、計算速度快,但在菌株間進化速率差異較大的情況下,可能得到錯誤的拓撲結構[15]。
1.4.2 最大簡約法 最大簡約法是一種將細菌的核苷酸序列位點視為形狀的方法,是一個比較所有可能的拓撲結構的過程。最大簡約法首先篩選出對細菌進化分析有用的核苷酸位點,然后統計每個位點的核苷酸最小替換數,進而以各位點替代數總和最小的進化樹作為最優樹[16]。相對于距離矩陣法,最大簡約法對信息的利用度更高,而相對于極大似然法和后驗概率法,該方法計算速度更快,可處理較多的插入、缺失序列。
1.4.3 極大似然法 極大似然法是利用進化模型和核苷酸序列,通過進化樹的分枝長度、拓撲結構、模型參數構建進化樹的方法。極大似然法通過選取合適的進化模型分析核苷酸序列,得到似然率最大的拓撲結構,再以其中最大似然率的拓撲結構構建的進化樹作為最優樹,應用極大似然法分析比較重要的問題時需要確定最合適的進化模型[17]。一般在選擇合理、正確的進化模型的情況下,極大似然法可以推導出很好的進化樹結果,但與最大簡約法相比,極大似然法很難在序列長度較短的情況下得到正確的拓撲結構。
1.4.4 后驗概率法 后驗概率法又被稱為貝葉斯推論法,該方法首先假定所有可能的進化樹是等概率的,然后計算出比對后序列的進化樹的后驗概率,進而將后驗概率最大的進化樹作為最優樹[18]。后驗概率法的優點在于不但具有數學和統計學基礎,同時還可以處理復雜的、接近實際情況的進化模型。與極大似然法相比,后驗概率法同樣應用廣泛,且可通過相同的數據信息,更低的計算量,得出與極大似然法一致的結論。與最大簡約法相比,后驗概率法能夠考慮更多的進化相關信息。
2 進化樹的構建
2.1 基于保守基因構建進化樹
2.1.1 基于核心基因組多位點序列分型(multilocussequence typing,MLST)構建進化樹 核心基因序列是重復且保守的核苷酸序列,在進化過程中可能發生富集、選擇和遺傳[19]。細菌的核心基因序列之間可以通過直接接觸及質粒、噬菌體或其他可移動遺傳元件(整合子、轉座子和插入序列等)的方式進行基因復制或基因交換,通過分析這些保守序列,可以分析不同細菌進化的差異以及菌株間的親緣關系[20]。MLST是近年來發展迅速的分子生物學分析方法,具有較高的分辨能力,可以通過多個管家基因450 bp左右的基因序列比較菌株等位基因的多態性,不同菌株對應不同的序列型,可以進行菌株進化和種群結構的研究[21]。核心基因組MLST可以使用微生物數百甚至數千個保守的等位基因進行基因分型,其分辨率遠高于傳統的MLST[22]。隨著NGS技術的廣泛應用,核心基因組MLST主要被應用在分子流行病學分析領域,且在該領域快速發展,已有研究采用核心基因組MLST方法分析我國即食食品中單核細胞增生李斯特菌的分子流行病學特征,結果表明核心基因組MLST能將不同譜系、血清群和克隆群的菌株明顯分開,共分為24個亞群,與克隆群基本保持一致[23]。基于全基因組測序的核心基因組MLST分辨能力強,可用于監測暴發性食源性疾病。但核心基因組MLST仍有一定的局限性,如缺乏對關系非常密切的菌株的分辨能力,且可分析菌株的種類有限[24]。2011年,SAHL等[25]對大腸埃希菌的MLST研究結果顯示,全基因組序列進化樹與MLST分型的結果并不一致。2014年,朱健銘等[26]對肺炎克雷伯菌進行分析,結果表明采用單個核苷酸序列進行細菌進化分析及采用MLST進行菌株親緣關系分析并不可靠,為了校正MLST的缺點,提高分辨率,他們采用管家基因和毒力基因聯合檢測的方法進行分析,發現根據核心基因組各位點序列圖譜構建的進化樹,與參與菌株相應序列進行比對,既可以準確地研究細菌遺傳進化關系,又可以確定菌株的種屬,對分析細菌親緣關系意義重大。
2.1.2 基于非編碼保守基因構建進化樹 非編碼保守DNA序列(conserved noncoding DNA sequences,CNS)是指細菌基因組中轉錄RNA但不能翻譯蛋白質或能調控其他基因的序列,是比較小的一段序列[27]。CNS在生物的進化中具有加工修飾RNA、調控轉錄和DNA結構等特殊的功能[28]。CNS不僅具有長度和頻率的物種特異性,還具有豐富性、廣闊性、保守性和功能性等特點,使其在微生物親緣關系的分析中極具潛力,如對耐輻射球菌與嗜熱菌親緣關系的分析[29]。但是如果要對CNS的功能進行正確、全面的理解和分析,還需要通過大量的實驗進行功能驗證。
2.2 基于單核苷酸多態性(single nucleotidepolymorphism,SNP)構建進化樹
SNP是指基因組中的單個堿基的突變引起的DNA序列多態性,有數量多、多態性豐富、遺傳穩定、易實現分析自動化的特點[30]。單個堿基的變異可以由顛換或轉換引起,也可以由插入或缺失引起,這些SNP位點可能影響基因的功能,引起性狀的改變,甚至導致疾病的發生,因此SNP是遺傳變異的重要依據,被廣泛應用于微生物的起源、進化及遷移等方面的研究。有學者為了解外源基因轉化沙漠寡營養細菌的進化與變異,通過生物學信息繪制了細菌的SNP系統發育樹,結果表明SNP數量最多的菌株進化速度最快[31]。SNP分析不僅提供了研究和理解基因突變的新方法,還能為微生物的鑒定及親緣關系分析提供依據。
2.3 基于基因拷貝數構建進化樹
基因拷貝數是某種基因或某段特定DNA序列在基因組中出現的數目,多拷貝基因廣泛存在于細菌中,而且多是可以移動的基因序列,分析基因拷貝數變異(jcopy number variation,CNV)是研究微生物進化、變異以及致病性的基礎[32]。CNV是基因組中的多核苷酸突變,是一種重要的遺傳變異,與研究較多的SNP相比,CNV涉及更多的堿基,覆蓋更大范圍的基因序列,在基因突變與細菌進化的研究上逐漸成為熱點和重點[30]。目前,全基因組CNV檢測的方法主要有芯片法和NGS技術等。全基因組CNV圖譜和更精確的參考基因組必將引領微生物基因組學研究熱潮,并進一步提高在全基因組范圍內探測基因組變異的準確性,對微生物親緣關系的分析有重大的指導意義。
3 基于其他特征基因構建進化樹
3.1 基于致病島構建進化樹
致病島又被稱為毒力島、適應島、生態島或共生島,是細菌基因組中可以編碼毒力因子的序列,與細菌的致病性密切相關,能夠在菌株間通過基因組水平轉移,可以使細菌在短期內發生形狀的改變,甚至產生新的變種,這種演變有助于細菌不斷適應環境[33]。很多病原菌都有致病島,如產腸毒素葡萄球菌、幽門螺桿菌、大腸埃希菌、沙門菌等[34-35]。基于致病島構建進化樹有助于理解細菌的進化和遷徙。有學者[36]研究了基于幽門螺桿菌CagPAI和Cag A基因構建的系統發育樹,驗證了拉丁美洲菌株的分群和聚類特點,以及與之相關的人群背景。隨著微生物耐藥性的增強和新型病原體的出現,鑒定致病菌也越來越重要。鑒定病原體的毒力基因并了解其從非致病性向致病性的進化,對于基礎科學和醫學研究都是一種挑戰。
3.2 基于規律成簇間隔的短回文重復序列(clustered regularly interspaced short palindromic repeats,CRISPR)構建進化樹
CRISPR在細菌基因組中由不連續的同向重復序列和插入其中的間隔序列組成,有針對噬菌體或質粒等外源基因的獲得性免疫作用,在細菌進化過程中保持結構的高度可變,CRISPR位點是研究細菌分型與進化的關鍵位點[37]。CRISPR通常由同向重復序列、間隔序列、前導序列以及CRISPR相關蛋白組成。cas基因與重復序列相互關聯,可使細菌協同進化[38]。CRISPR位點會隨著細菌的進化不斷出現新間隔序列的插入以及舊間隔序列的丟失,這種現象是導致細菌基因組進化速度快的重要原因之一,因此CRISPR位點具有很復雜的多態性[39]。CRISPR位點在細菌中的多態性不僅能夠反映細菌與環境相互作用的關系,還能夠記錄細菌在進化過程中的生態學和地理學信息,如根據CRISPR位點的間隔序列在沙門菌中排列的差異,可以判斷不同菌株間的親緣關系,并進行溯源分析;根據醋酸菌的重復序列構建進化樹,可將不同屬的菌種進行分類[40]。有研究應用CRISPR序列分型分析病原體的暴發流行,如鼠疫耶爾森菌和腸炎沙門菌亞種的分群[40-41]。此外,CRISPR還可以提供與微生物表型相關的重要信息,如腸球菌耐藥基因序列和化膿性鏈球菌基因組中的前噬菌體等,這些信息都反映了CRISPR在調控基因水平轉移、細菌適應環境及細菌進化中的作用[42]。因此,應用CRISPR構建進化樹分析細菌親緣關系的優點在于:基因分型分辨率較高、操作簡單、重復性好、結果數字化、便于不同實驗室結果的比對等[43]。但目前仍然存在一些需要解決的問題,如數據不夠充分、數據庫不夠健全、細菌之間的分型標準不夠完善等,在未來,構建標準化的CRISPR數據庫具有十分重要的意義。
4 總結和展望
基因分析技術的飛速發展促進了生物學諸多領域的發展,對認識細菌基因組與進化的關系有深遠的影響。細菌基因組具有多樣性和規律性特征,面對海量的基因組信息,挖掘有效信息,構建合適的進化樹是十分重要的。有效地構建進化樹應該具備2個條件:首先,必須基于一種合適的進化方式,能反映或解釋進化事件;其次,應該覆蓋更多的基因組信息[44]。為更好地鑒定細菌類型以及明確不同菌群之間的親緣關系,結合不同基因組序列構建細菌進化樹,將有效幫助解決細菌進化中許多懸而未決的問題[45]。
本文簡述了目前常見的用于構建細菌進化樹的方法,但是可以用來進行細菌親緣關系分析而構建進化樹的方法不限于文中所述,挖掘更多有價值的基因標志物將是很有前景的研究,也是探究細菌進化史的必由之路。
猜你喜歡
現代畜牧科技(2021年9期)2021-10-13 06:39:14
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
當代經濟研究(2016年5期)2016-12-01 03:12:05
現代農業(2016年5期)2016-02-28 18:42:46
出版與印刷(2016年3期)2016-02-02 01:20:11
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
華北水利水電大學學報(社會科學版)(2014年3期)2014-04-16 04:38:31
終身教育研究(2014年5期)2014-02-28 01:23:06
