高效液相色譜-串聯質譜測定紡織品中2種樟腦衍生物類光穩定劑
2021-01-12 08:09:32連秋燕朱峰施點望林麗云邱尚仁
福建輕紡 2021年1期
連秋燕,朱峰,施點望,林麗云,邱尚仁
[1.國家紡織服裝產品質量監督檢驗中心(福建),福建 福州 350026;2.福建省紡織產品檢測技術重點實驗室,福建 福州 350026]
3-亞芐基樟腦(3-BC,CAS:15087-24-8)和4-甲基亞芐基樟腦(4-MBC,CAS:36861-47-9)等樟腦衍生物類化合物具有六環結構,通常有很高的紫外線吸收效率,具有強UVA段吸收,被添加到紡織品[1]、塑料、化妝品、護發品等工業產品,也用作涂料、顏料、涂層等的光穩定劑,以延緩產品老化[2-3]。紡織品經抗紫外整理后,可以減少織物泛黃或變脆的現象,而且可以保護人體皮膚少受紫外線的傷害,因此,在抗紫外織物的市場日益擴大的情況下上述物質被大量使用[4]。
研究表明,3-亞芐基樟腦和4-甲基亞芐基樟腦具有胚胎毒性,存在內分泌干擾特性,對人體有長期且不可逆的影響[5-6]。來自德國提交給歐盟的技術報告顯示:4-甲基亞芐基樟腦和3-苯亞甲基樟腦的雌性激素干擾模式類似于對特辛基苯酚(CAS:140-66-9)。
2007年,我國《化妝品衛生規范》將3-亞芐基樟腦和4-甲基亞芐基樟腦列為限用防曬劑,限定了其加入量。國際上對化妝品中防曬劑的使用量和種類有嚴格的管理和限制[7-8],2016年2月29日,歐洲化學品管理局(ECHA)將4-甲基亞芐基樟腦、3-苯亞甲基樟腦等4種物質列入高關注物質(SVHC)咨詢清單,對其提起公眾咨詢。2018年12月18日,歐盟在其官方公報(OJ)上公布了一項決議(E U)2018/2013,正式將3-亞芐基樟腦確認為高度關注物質(SVHC)。
對3-亞芐基樟腦(3-BC)和4-甲基亞芐基樟腦(4-MBC)檢測的研究主要是在化妝品中防曬劑的測定[9-12],因為目前為止,國內外還未發現紡織品中3-BC和4-MBC檢測方法的相關研究報道,因此,開展此項研究具有重大的意義。
本文建立了紡織品中3-BC和4-MBC的(LCMS/MS)分析方法[13]。該方法采用超聲萃取,高效液相色譜分離,串聯質譜電噴霧離子源(ESI)電離,正離子多反應監測模式(MRM)進行定性和定量。方法具有簡單、快速、靈敏、準確的特點,用于紡織樣品中3-BC和4-MBC的檢測分析,為嚴格質量管理監控提供必要的技術支持。2種樟腦衍生物的化學信息見表2。
1 實驗部分
1.1 儀器與試劑
1290 Infinity高效液相色譜-6410B串聯質譜儀:配有2級質量分析檢測(HPLC-MS/MS),美國Agilent公司;97043-942超聲波發生器,美國VWR公司;提取器:具密封塞提取瓶;1 mL一次性注射器;0.22 μm尼龍過濾頭。
甲醇、乙腈、正己烷、乙酸乙酯、二氯甲烷、丙酮均為色譜純。
甲酸銨,純度≥98.0%(德國CNW科技公司); 標準品:3-亞芐基樟腦(3-BC)純度為 99.6%、4-甲基亞芐基樟腦(4-MBC):純度為97%(德國CNW科技公司)。
1.2 標準溶液的配制
精密稱取標準品3-BC和4-MBC,用甲醇配制的標準儲備液(1000 mg/L),0~4 ℃保存。根據需要逐級稀釋至相應濃度。
1.3 樣品前處理
稱取1.0 g已剪碎混勻的試樣(精確至0.001 g),置于具密封塞提取瓶中,加入10 mL乙腈溶液,超聲波50 ℃中超聲振蕩30 min。過0.22 μm濾膜,濾液用HPLC-MS/MS檢測。
1.4 色譜和質譜條件
1.4.1 HPLC條件
色譜柱:美國Agilent公司ZORBAX SB C18柱(2.1 mm×150 mm,3.5 μm;);柱溫:35 ℃;流動相A:甲醇;流動相B:0.01 mol/L甲酸胺水溶液。
梯度洗脫條件:0~2 min,85% A~90% A;2~3.5 min,90% A~100% A;3.5~5 min,100% A;5~6 min,100% A~85 % A;6~8 min,85% A。
進樣體積:5.0 μL;流速:0.2 mL/min。

表1 2種樟腦衍生物的化學信息

表2 2種化合物的多反應監測質譜參數和保留時間
1.4.2 MS/MS條件
檢測方式:多反應監測(MRM)模式;正離子掃描;離子源溫度:350 ℃;電噴霧電壓:4000 V;霧化氣壓力:300 kPa;干燥氣流速:12 L/min。其他參數如表2所示。
2 結果與討論
2.1 色譜柱的優化
3-亞芐基樟腦和4-甲基亞芐基樟腦結構相似,分子量接近,要使他們在色譜上實現分離,色譜柱的選擇是關鍵。因此實驗考察了不同型號規格,不同長度的5種色譜柱對3-亞芐基樟腦和4-甲基亞芐基樟腦分離效果,實驗結果表明:Zorbax SB-C18柱(2.1 mm×150 mm,3.5 μm)可以實現2個化合物的有效分離。
2.2 流動相的優化
實驗對甲醇-水溶液和乙腈-水溶液進行比較分析。發現:2個體系對2個化合物的分離效果差異不大,于是選擇了2個化合物響應值較高的甲醇體系,且考察了在水相中添加0.1 %甲酸、0.01 mol/L甲酸銨、0.01 mol/L乙酸銨對2個化合物分離的影響,結果表明用0.01 mol/L甲酸銨水溶液為流動相時,2個化合物的分離度更好、靈敏度更高,因此選擇甲醇-0.01 mol/L甲酸銨水溶液為流動相。

圖1 2種化合物的MRM圖。
實驗對流動相梯度洗脫條件、色譜柱溫和流動相流速進行了優化,結果如1.4.1中的HPLC條件,使得2種化合物實現基線分離,獲得較高的響應值。
2.3 質譜條件的優化
分別用正、負離子掃描3-亞芐基樟腦和4-甲基亞芐基樟腦,結果表明:3-亞芐基樟腦和4-甲基亞芐基樟腦在正離子掃描模式分子峰較明顯,在負離子模式下質譜響應很弱,此選擇正離子掃描模式。實驗進一步優化了母離子和子離子,對質譜條件進行了優化選擇,確定最佳的質譜參數如1.4.2所列。最佳質譜條件下,得出的2個化合物的MRM圖如圖1所示。
2.4 前處理方法的優化
實驗采用超聲波提取方法,比較了乙腈、甲醇、乙酸乙酯、正己烷、二氯甲烷對3-亞芐基樟腦和4-甲基亞芐基樟腦的提取效率,結果如圖2所示,乙腈對2種化合物的提取效率最高。實驗選擇乙腈作為提取溶劑,并進一步考察了提取溶劑體積對提取效率的影響,同時優化了超聲溫度,超聲時間,得出最佳的前處理實驗條件如1.3所列。
2.5 方法的線性關系和檢出限
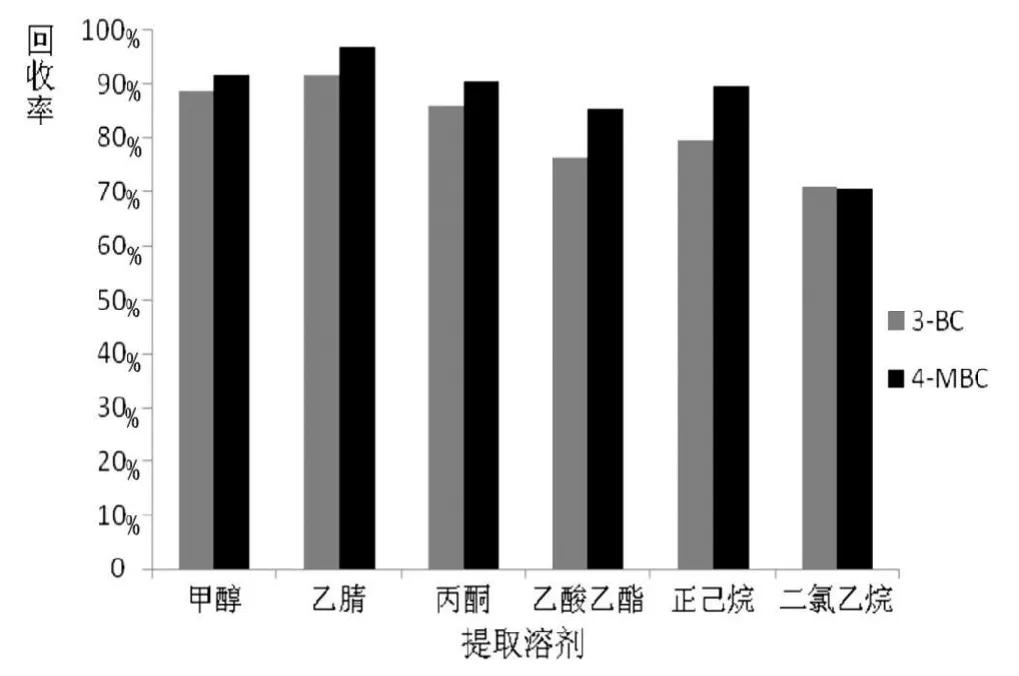
圖 2 提取溶劑對2種化合物提取效率的影響(n=3)
配置一定濃度范圍的3-亞芐基樟腦和4-甲基亞芐基樟腦標準溶液,在優化的實驗條件下進行測定,以化合物的質量濃度(c,μg/L )為橫坐標,以化合物在MRM模式下定量離子的峰面積為縱坐標(y),繪制標準曲線,得到線性回歸方程和相關系數(R2);分別按信噪比(S/N =3)和S/N =10時的質量濃度確定方法的檢出限(LOD)和定量限(LOQ),結果如表3。

表3 2個化合物的線性方程、檢出限和測定低限
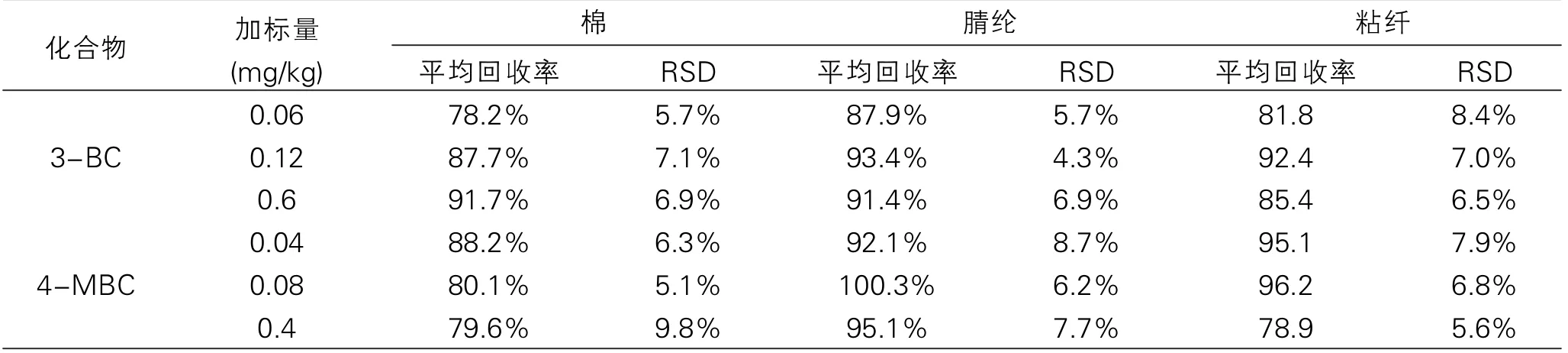
表4 方法的回收率和相對標準偏差(n=6)
2.6 回收率與精密度
實驗選取了棉、腈綸、粘纖等陰性樣品,在最佳前處理條件下,進行6次的加標回收率實驗,實驗添加的分別是測定低限、2倍測定低限、10倍測定低限3個濃度水平的目標化合物。
在優化的色譜條件下測定。實驗結果(表4):3-亞芐基樟腦和4-甲基亞芐基樟腦的平均回收率為78.2%~100.3%,相對標準偏差為4.3%~9.8%,說明該方法的重現性好,精密度高,能滿足紡織品中2種樟腦類衍生化合物紫外吸收劑的測定要求。
3 結論
利用高效液相色譜-串聯質譜建立了紡織品中3-亞芐基樟腦和4-甲基亞芐基樟腦2種樟腦類衍生化合物的檢測方法,通過優化提取溶劑等前處理條件和質譜檢測條件,在短時間內實現2種化合物的有效分離,該方法簡便、快速、靈敏、準確,適用于紡織品中2種樟腦衍生類化合物的測定,為嚴格質量管理監控提供必要的技術支持。
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
房地產導刊(2022年5期)2022-06-01 06:20:14
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
建材發展導向(2021年12期)2021-07-22 08:06:48
建材發展導向(2021年7期)2021-07-16 07:07:52
中學生數理化(高中版.高二數學)(2021年12期)2021-04-26 07:43:48
中學生數理化(高中版.高考數學)(2021年12期)2021-03-08 01:28:50
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
