AMP激活蛋白激酶對慢性心力衰竭進展影響的實驗研究
2021-01-15 08:45:14金愛萍張倩榕
陜西醫學雜志 2021年1期
金愛萍,李 蔚,李 冰,張倩榕
(西安交通大學第二附屬醫院老年心血管科,陜西 西安710004)
AMP激活蛋白激酶(Mammalian AMP-activated protein kinase,AMPK)是一種絲氨酸/蘇氨酸蛋白激酶,包含催化α亞單位和調節β和γ亞單位[1]。AMPK對心臟肥大和心力衰竭具有保護作用,可通過阻斷各種信號通路,包括活化T細胞核因子(Activated T cells nuclear factor,NFAT)、絲裂原活化蛋白激酶(Mitogen-activated protein kinase,MAPK)、核因子κB(Nuclear factor kappa-B,NF-κB)、叉頭轉錄因子(Forkhead box O,FOXO)和肌肉特異性環指蛋白1(Muscle specific ring finger protein 1,MuRF1)的核因子減輕由壓力超負荷或去氧腎上腺素(Phenylephrine,PE)引起心臟肥大[2]。已有研究顯示[3],抑制自噬反應和鈣離子/鈣調素依賴激酶β-(CAMKK-β)-AMPK信號通路活性能夠延緩心臟肥大進程;激活AMPK有助于提高心肌細胞肥大代償能力、改善心肌缺血缺氧狀態。此外AMPK誘導自噬反應能夠干擾心臟肥大進程,阻止心力衰竭病情發展;但自噬調節異常亦可能是導致心力衰竭發生發展。目前對于AMPK能否通過調節自噬來達到保護心力衰竭患者心臟功能作用尚存在爭議[4]。本次研究通過探討AMPK激活對慢性心力衰竭病情的影響及作用機制,旨在尋找潛在有效治療靶點,現報告如下。
1 材料和方法
1.1 實驗材料 GAPDH、Beclin-1、Pho核糖體蛋白S6激酶(P70S6K)(T389)、P70S6K、Pho-4E結合蛋白1(4EBP1)(T37/46)、4EBP1、Pho-AKT(Ser308)、Pho-AKT(Ser473)及AKT均由Cell Signaling Technology公司提供;Lc3b由Novus公司提供;α肌動蛋白和P62由Sigma公司提供;Ampk激動劑(AICAR)由Toronto chemicals公司提供。
1.2 實驗方法
1.2.1 實驗動物、造模及給藥方法:選擇8~10周齡雄性C57BL/6J小鼠共40只,體重24~26 g,均由我校實驗動物中心提供;小鼠腹腔注射1.5%戊巴比妥麻醉后行主動脈縮窄術,4周后建立心力衰竭模型;其中A組為生理鹽水(NS)注射+假手術(sham),B組為AICAR注射+假手術(sham),C組為NS注射+行主動脈縮窄手術,D組為AICAR注射+行主動脈縮窄手術。
1.2.2 超聲心動圖檢查:檢查前小鼠吸入1.5%~2%異氟醚;采用Visual Sonics公司生產Vevo 2000型動物專用超聲診斷儀,探頭頻率為30 MHz;小鼠取左側臥位或仰臥位,并在剃毛區均勻涂抹耦合劑,選取左心室乳頭肌短軸切面測量。
1.2.3 組織取材測量:稱量小鼠體重并記錄,頸椎脫臼法迅速處死小鼠,沿胸骨正中向頸部剪開皮膚及橫隔,充分暴露心臟和肺臟;剪下心臟后輕輕擠壓心腔內血液,修剪心臟周圍多余部分,記錄心臟重量。取出小鼠肺臟,修剪后濾紙吸干,稱重并記錄。剪開小鼠后肢脛骨處皮膚,測量并記錄脛骨長度;計算心重量與體重的比值(HW/BW),肺重量與體重的比值(LW/BW)以及心重與脛骨長度的比值(HW/TL)。
1.2.4 Western blotting檢測:采用細胞溶解緩沖液+PMSF+蛋白酶抑制劑混合液提取左心室組織蛋白質;蛋白濃度檢測采用Bio-Rad 蛋白分析試劑盒;Western blotting操作完成后通過Quality One軟件完成定量分析。
1.2.5 熒光共焦顯微鏡檢查:制備心臟組織冰凍切片(厚度5 mm),干燥后采用PBS洗滌并在過氧化氫(0.3%)溶液中孵育30 min;制備樣品在室溫下以BSA(10%)封閉1 h,一抗孵育,再采用熒光二抗洗滌孵育;采用Zeiss LSM 600型共聚焦顯微鏡系統成像。
1.2.6 透射電子顯微鏡檢查:制備1~2 mm3組織塊,浸泡于2%多聚甲醛和2.5%戊二醛4 h以上,1%OsO4中固定1 h;采用超顯微切片機將組織制備成60~80 nm切片,放置于透射電鏡網格并行檸檬酸鉛染色,80 kV電壓下采用FEI公司雙透射電子顯微鏡成像。

2 結 果
2.1 兩組心臟功能相關實驗指標比較 C組HW/BW、LW/BW、HW/TL、肺重量/脛骨長度比(LW/TL)、左心室舒張末期內徑(LVEDd)及左心室收縮末期內徑(LVESd)水平均顯著高于A、B組(P<0.05);C組射血分數(Ejection fraction,EF)水平顯著低于A、B組(P<0.05);D組HW/BW、LW/BW、HW/TL、LW/TL、LVEDd及LVESd水平均顯著高于A、B組,但低于C組(P<0.05);D組EF水平顯著低于A、B組,但高于C組(P<0.05)。見表1。
2.2 AMPK對心力衰竭模型動物mTOR信號通路的影響 B組術后4EBP1、P70S6K及AKT磷酸化水平顯著高于A組(P<0.05);D組術后4EBP1、P70S6K及AKT(Thr308)磷酸化水平顯著低于C組(P<0.05);D組術后AKT(Ser473)磷酸化水平顯著高于C組(P<0.05)。
2.3 AMPK對于慢性心力衰竭患者自噬的抑制作用 D組術后LC3B-Ⅱ/LC3B-Ⅰ和LC3B-Ⅱ/GAPDH比值顯著低于C組,P62表達顯著高于C組(P<0.05);C組和D組Beclin-1水平比較差異無統計學意義(P>0.05)。C組和D組心臟LC3b蛋白表達量顯著高于A組、B組(P<0.05);D組心臟LC3b蛋白表達量顯著低于C組(P<0.05);同時B組心臟LC3b蛋白表達量顯著高于A組(P<0.05),見圖1。
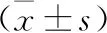
表1 兩組心臟功能相關實驗指標比較

圖1 AICAR給藥后心臟自噬現象(熒光染色,×200倍)
3 討 論
自噬反應是維持心肌形態及心臟功能重要組成部分之一;已有研究顯示[5],脂聯素可刺激自噬反應,抑制心肌細胞肥大,從而改善心臟功能;同時激活Sirt3可在調節自噬反應同時改善心肌肥大病情。但亦有報道認為[6-7],心力衰竭和過度活躍自噬反應密切相關。早期實驗研究證實[8],AMPK可通過激活自噬有效抑制心臟肥大發生,但對于自噬反應過度激活及調節紊亂亦可加速心力衰竭病情發展。基于以上數據,本次研究目的在于探索AMPK是否可通過調節自噬作用達到改善心力衰竭病情作用。
以往實驗研究證實[9],心臟肥大時AMPK被激活并能夠延緩心力衰竭病情進展,提示AMPK在心力衰竭病變發生前即已開始誘導自噬反應。同時在以往建模過程中往往行主動脈縮窄手術4周后完成慢性心力衰竭模型建立,再于心力衰竭階段給予AICAR給藥,此時自噬反應已被過度激活[10]。而本次研究結果中,AMPK可通過下調自噬反應來促進心力衰竭模型動物心臟功能恢復;同時AMPK增強行假手術組模型動物自噬反應,但心臟功能仍正常。筆者推測AMPK可通過上調心肌肥大時自噬反應和下調心力衰竭時過度自噬來達到改善心臟功能的作用。
mTOR包括兩個功能不同復合物,即mTORC1和mTORC2;其中mTORC1由mTORC、Raptor、Mlst8及Pras40組成,具有調節蛋白質合成、細胞增殖、代謝及自噬等多方面作用;而mTORC2則由mTORC、Rictor、Mlst8、Protor(prr5)及Sin1組成,主要用于控制細胞存活及調節極性[11-12]。已有研究顯示[13],損傷組織細胞往往失去增殖潛能和蛋白沉積調節作用,細胞內積聚大量毒性碎片,破壞正常細胞代謝進程,最終誘導蛋白毒性應激反應發生。另有研究表明[14-15],心肌細胞肥大及凋亡可被mTORC1受體顯著抑制,可能機制為清除蛋白毒性應激源和高炎癥性衰老表型組織細胞。AMPK是mTORC1重要調節因子之一,AMPK-mTORC1通路可參與到心力衰竭自噬及能量代謝調節過程中[16]。本次研究結果中,AMPK可有效抑制衰竭心臟自噬活性,下調mTORC1表達。目前并無特異性mTORC1抑制劑能夠在抑制TORC1功能同時不影響其他生理作用,故mTORC1對于心肌肥大影響無法完全歸因于其對于自噬反應調節作用[17-18]。此外糖尿病心力衰竭患者中可見mTORC活性下降,但AMPK活性未見明顯變化[19]。故筆者認為AMPK激活后mTORC1下調可能未參與到心力衰竭自噬調節過程。
mTORC2對于自噬反應影響仍存在爭議;以往研究顯示[20],AMPK通過調節mTORC1相關自噬反應以抑制心臟肥大。但本次研究結果中,AMPK在下調模型動物心臟自噬活性同時激活mTORC2,說明AMPK可通過刺激mTORC2活性以調節過度激活自噬反應,同時Stim1亦可通過抑制mTORC2以加快心力衰竭病情進展;已有研究證實[21],mTORC2可對心力衰竭模型小鼠心臟收縮功能進行調節;筆者推測通過mTORC2下調自噬反應及AMPK激活可延緩心力衰竭病情進展。
綜上所述,慢性心力衰竭患者體內AMPK激活可通過上調mTORC2表達、抑制心臟自噬活性,從而達到有效改善心臟功能的作用。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
中華詩詞(2022年6期)2022-12-31 06:41:24
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
學苑創造·A版(2020年9期)2020-10-13 09:41:02
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
中國科技論壇(2017年7期)2017-07-25 08:49:53
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
中國中醫藥現代遠程教育(2014年22期)2014-03-01 04:32:55
