
金屬有機骨架材料在金屬鋰電池界面的應用
2021-03-05 12:30:22孫宇恒高銘達李慧徐麗薛晴王欣然白瑩吳川
物理化學學報 2021年1期
孫宇恒 ,高銘達 ,李慧 ,徐麗 ,薛晴 ,王欣然 ,白瑩 ,吳川 ,3,*
1環境科學與工程北京市重點實驗室,北京理工大學材料學院,北京 100081
2先進輸電技術國家重點實驗室(全球能源互聯網研究院有限公司),北京 102209
3北京電動汽車協同創新中心,北京 100081
1 引言
人類對化石燃料的過度依賴造成全球能源短缺、二氧化碳排放超標、有害氣體大量生成等環境問題。為了解決上述問題,各國推動了清潔、可再生能源的發展和規模化利用。由于可再生能源(風能、核能、水能等)存在地域分布不均、晝夜波動較大等缺點,高效的能源存儲是規模化利用可再生能源的前提。其中,高比能二次電池具備較高的能量密度和工作電壓,以及無記憶效應等諸多優點,已經廣泛的應用到可移動電子終端設備、電動汽車、智能電網等領域1。
然而,高比能二次電池的界面問題阻礙了能源行業的進一步發展,這些問題包括電極界面處電解液副反應、溶劑共嵌入、鋰枝晶以及死鋰生成等,其加劇了活性物質在循環過程中的損失,并且阻礙了固體電解質界面(Solid electrolyte interphase,SEI)的生成與穩定。界面問題的存在既影響了電池的性能又降低了電池的安全性。特別是在高比能鋰金屬電池體系中,由于界面的不均一性和高反應活性,副反應持續發生,界面SEI膜存在缺陷,導致鋰離子無法在鋰金屬表面均勻成核,形成鋰枝晶,并進一步產生庫倫效率降低、鋰負極體積增加、電池內阻增大等問題2;在金屬鋰固態電池體系中,固態電解質與電極材料的界面接觸不佳,會產生較大的接觸阻抗,降低離子傳輸效率和電化學反應可逆性,同時空間電荷區的出現也為加劇了鋰枝晶的生長3;此外,一些典型的金屬鋰電池問題,如:鋰硫電池的多硫化物穿梭效應、高電壓正極材料的過渡金屬溶出等,都與界面電化學過程息息相關4。因此,強化界面穩定性和保護功能,是改善金屬鋰負極界面穩定性、高比能二次電池電化學性能的關鍵。
為解決高比能二次電池的界面問題,科研人員已經研究了諸多類型的材料,近年來較為熱門的金屬有機骨架(Metal organic framework,MOF)材料和共價有機骨架(Covalent organic framework,COF)材料也被廣泛的研究應用于界面保護。同為多孔骨架材料,MOF和COF都有著獨特的孔道結構以及大的比表面積,良好的功能化能力。相對而言,COF材料是晶態的有機多孔聚合物,由共價鍵連接,密度低、結構穩定,已經有研究將其應用于金屬鋰負極的人造SEI膜5、固態聚合物電解質6以及鋰硫電池的正極材料7等,然而由于其合成較為困難、固有的離子電導率較低等因素,目前其對于電池界面保護的研究較少。
金屬有機骨架材料又稱多孔配位聚合物材料,是由金屬離子/簇作為節點,有機配體作為連接體,通過配位形成的多孔晶體材料。MOF材料具有規整且可調控的孔道結構、較大的比表面積和孔隙率,以及配位不飽和的金屬位點(也稱為開放金屬位點)8,9。通過對金屬節點和有機連接體的合理設計,MOF可實現高效的氣體吸附10-12、分離純化13、催化14-16、納米醫學17,18等應用。在高比能金屬鋰電池研究方面,MOF的規整孔結構能提供良好的鋰離子擴散通道,并改善界面反應不穩定以及離子傳導不佳的問題。作為儲鋰基體或者人工SEI膜材料,MOF的多孔性使得電極界面離子流均勻化,并抑制鋰枝晶的生成。在鋰硫電池中,MOF作為有效的多硫化物捕集器,其開放的金屬位點和功能化的連接物與多硫化物發生化學吸附,從而改善界面反應性能19。在鋰-氧氣電池中,MOF能夠作為高效界面反應催化劑,在開放金屬位點進行氧還原反應,從而有效地穩定界面反應,提高容量與能量轉換效率。在固態電池中,MOF作為全固態電解質的鋰離子導體,其中鋰離子可沿著開放的金屬位點快速移動。因此,本文綜述了近年來MOF材料在解決新型高比能鋰金屬電池界面問題中的最新研究進展(如:金屬鋰電池、鋰硫電池、鋰空電池和固態電池等),以及MOF材料抑制正負極界面問題和改善隔膜界面特性中的應用。
2 MOF材料的結構特點
2.1 MOF均勻孔道結構與界面調控功能
MOF材料具有高孔隙率、高比表面積、孔結構可調控等結構優勢。早在1999年,Li等20對MOF-5的研究發現,其三維骨架結構中未被占據的晶體體積約為80%,可以有效結合客體分子的體積約占MOF材料的55%-61%,從而為氣體儲存提供了巨大的可用潛力。2006年,Li等21首次將鋅基MOF(MOF-177)應用于鋰儲存,為其在二次電池領域中的應用提供了重要的指導意義。MOF材料擁有較大的比表面積(1000-10000 m2·g-1)22,這種高孔隙率的結構能夠改善離子在MOF材料體相中的擴散動力學,從而提高電極的離子電導率以及電池的倍率性能23。
MOF材料通常可通過水熱-溶劑熱法24、微波輔助法25、電化學法26、噴霧干燥法27等方法合成。根據配合物中所含配體的不同,MOF主要包括含氮雜環類有機配體MOF、有機羧酸類配體MOF、含氮雜環與羧酸混合配體MOF28-35。在制備含氮雜環類有機配體MOF材料時,多采用吡啶類、咪唑類、2,2’-聯吡啶、4,4’-聯吡啶等中性配體,在這類材料中配體與金屬離子通過π鍵以及氫鍵形成多維結構,鍵合力弱結構易分解,其中較為常見的材料有類沸石咪唑酯骨架系列材料(ZIF)28,29,Coordination pillared-layer (CPL)系列材料30,31等;另一方面,采用羧基配體可以形成強配位鍵并構建穩定的大孔徑材料,較為常見的材料有Materials of Institut Lavoisier (MIL)系列材料32、Porous Coordination Network (PCN)系列材料33、奧斯陸大學系列材料(Universitetet i Oslo,UiOs)34等。為了追求更加優異的性能,羧酸類與含氮雜環類配合物復合的MOF材料,同時具備了骨架的穩定性和高維結構的優點,代表性的材料有架狀金屬有機骨架材料(IRMOF)35。如圖1a-f所示,根據配合物中所含中心金屬離子的種類不同,MOF材料又可劃分為過渡金屬型、稀土金屬型、堿金屬型,以及在此基礎上構建的多金屬MOF材料等。調控金屬節點和有機連接體的類型,可調控MOF孔道結構的大小。如圖1g-i所示,以苯二甲酸(BDC)、BDCNH2和BDC-NHCOCH3配體構成的UiO-66、UiO-66-NH2和UiO-66-NHCOCH3型MOF材料,其孔徑大小從1.18 nm縮減到0.44 nm36。
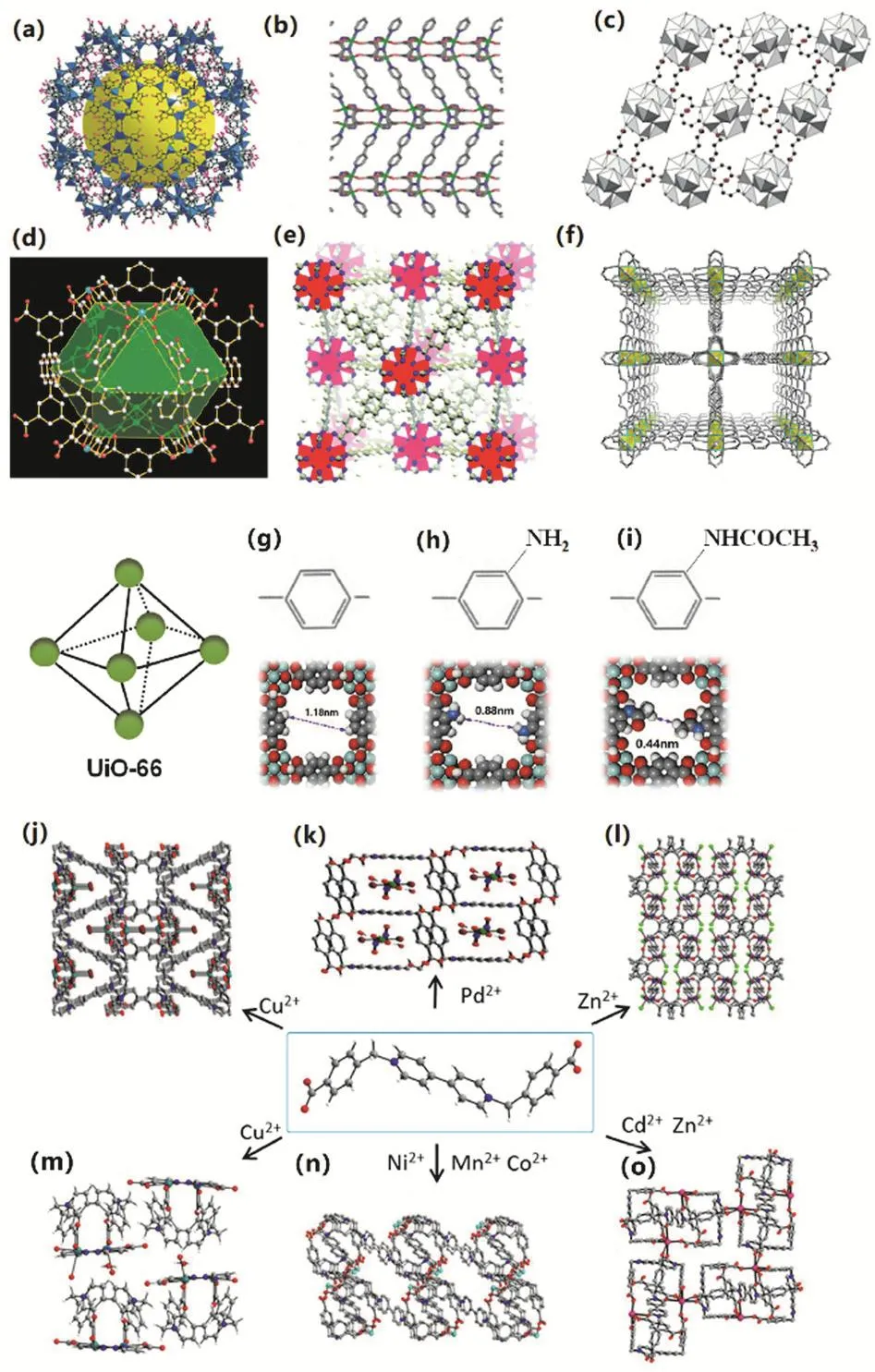
圖1 不同配基、中心離子組成的MOF材料的孔結構特性及變化規律Fig. 1 The porosity and structural changes of MOF derived from different organic ligands and central ions.
另一方面,由于中心原子的空軌道數各不相同,不同中心原子參與配位成鍵不同,因此,不同配位金屬所形成的孔道結構略有不同37。二價金屬具有較多的3d軌道,有利于形成多種配位構型。如圖1j-l所示,以配體2-氨基苯甲酸(2-abaH)38,不同金屬節點(Ca2+、Sr2+、Ba2+),可構建出不同結構和孔特性的MOF材料,其金屬原子的間距分別為0.4699、0.3917、0.4316 nm。Aulakh等39以1,1’-雙(4-羧芐基)-4,4’-聯吡啶二溴化物(H2LBr2)為配體,分別與Cu、Mn、Ni、Cd、Zn和Pd過渡元素通過自組裝反應形成了九種不同的兩性MOF材料。如圖1(j-o)所示,其主要包含六種結構:{CuBr(L)·(OH)·7H2O}n(圖1j),[Pd(HL)(Br)2(NO2)2(OH2)2] (圖1k),{[ZnCl2(L)0.5]·0.33H2O}n(圖1l),{[Cu2(3-pzc)2(L)(OH2)]·5H2O}n(圖1m),{[M4(L)6(OH2)12]·2Br·3(bdc)·33H2O}n(圖1n,其中M分別為Mn、Co和Ni和{[M(bdc)(L)1.5·9H2O]}n(圖1o,M分別為Cd和Zn,L為配體中的長鏈結構,bdc是1,4-苯二甲酸,pzc是3-吡唑羧酸鹽)。由于金屬離子的不同,MOF材料的孔徑不僅產生了差異并且結構也產生了變化。另外值得注意的是,在合成{[Cu2(3-pzc)2(L)(OH2)]·5H2O}n(圖1m)時額外加入了吡唑-3-羧酸,使得其與{CuBr(L)·(OH)·7H2O}n(圖1j)兩種MOF材料在配體與金屬離子相同的情況下結構仍然產生了變化。類似的,{[ZnCl2(L)0.5]·0.33H2O}n(圖1l)在合成時采用的是ZnCl2而{[Zn(bdc)(L)1.5·9H2O]}n(圖1o)在合 成時使用了Zn(CH3COO)2·2H2O,因此這兩種MOF的結構也不同。
2.2 MOF材料的金屬位點和功能性官能團
MOF的固有活性位點包括配位不飽和金屬位點和功能性有機位點(橋聯配體中的官能團),這些獨特的活性位點為穩定金屬鋰電池界面提供豐富的作用靶點。Chen等40通過單晶X射線衍射驗證了MOF-11中的開放金屬位點。在初始合成的MOF中,金屬離子通常都處于完全配位飽和的狀態,即對于過渡金屬離子而言配位數為4或6,如圖2a所示,開放金屬位點主要是通過在MOF結構中引入不穩定的客體分子(如丙酮或甲醇),參與金屬配位。在激活過程中,這些客體分子可以被熱移除,而不會導致結構坍塌,從而產生開放的金屬位點41,42。這些開放的金屬位點可以選擇性地與客體分子相互作用。Fu等43通過甲醇的加入來調控MOF中OMs的配位狀態,從而使MOFs獲得對極性分子良好的吸附能力。針對金屬鋰電池界面的復雜電化學反應,MOF配位不飽和的金屬位點也能夠有效地吸附反應副產物,如鋰硫電池的中間產物多硫化物,從而起到封裝、固定、轉化副產物,穩定界面電化學性能的作用44。對于催化性能的提升,Wu等45將MOF成功應用于鋰氧電池中,其均勻通道中易于進入的OMs增加了孔隙中O2分子的數量,有效地促進了反應的進行。Zhang等46利用靜電紡絲技術制備了UiO-66和聚乙烯醇(PVA)的復合隔膜,如圖2b所示,MOF中的OMs可以與孔道中的陰離子絡合,釋放鋰離子并顯著提高了鋰離子遷移數(0.79)和離子電導率,保證了快速有效的鋰離子傳輸。值得注意的是,MOF中的金屬位點所具有的氧化還原活性使其可以被用作“轉化型”電極的電極材料。Peng等47證實了合成的三苯氨基MOF中的Cu2+位點具有氧化還原活性并用作鋰離子電池正極材料,具有4.3 V的工作電壓,然而根據電化學測試結果,發現由于MOF中羧基氧原子限制了鋰離子的傳導,導致了對Cu+和Cu2+的不完全氧化還原反應,使整個系統的結構存在不穩定現象。合理的金屬位點選擇與結構設計以及添加劑的使用可以更有效的利用MOF中金屬位點的氧化還原活性。與此同時,合理的選擇設計功能化的有機連接體,能起到捕獲、催化客體分子的作用。Zhong等48通過溶劑熱法,構建了以鈷雙(二羥基)配合物(Co-O4)為鍵的銅酞菁基二維共軛MOF材料。原位拉曼光譜證實了在氧還原反應過程中,連接體中的Co-O單元與氧中間體存在相互作用,驗證了Co-O單元具有催化活性,獲得了金屬空氣電池94 mW·cm-2的功率密度和700 mAh·g-1的質量比容量。
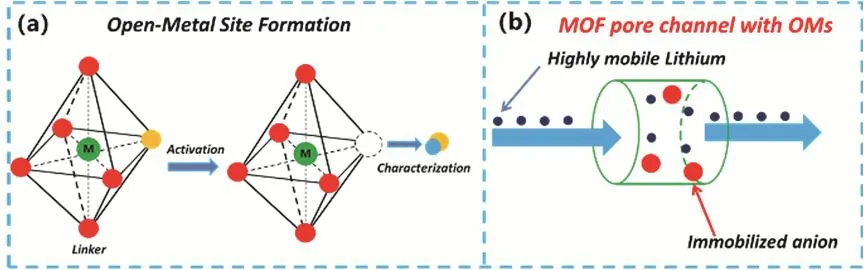
圖2 (a)配位不飽和金屬位點的產生42;(b)復合隔膜中OMs的功能示意圖46Fig. 2 (a) Generation of coordinated unsaturated metal sites 42, (b) function of OMs in separator 46.
2.3 MOF衍生材料的納微結構調控
MOF可通過煅燒等方式,轉化為多孔的碳材料49、金屬氧化物50,51、金屬氧化物-碳復合材料52和金屬納米粒子@MOF復合材料等53。其衍生材料保持了多孔結構的特性,同時具備納米尺寸、高比表面積、復合材料等界面調控優勢。研究人員借助不同的合成路徑和模板,獲得了不同維度的MOF衍生納米材料54。如圖3所示,MOF材料可原位生長在一維納米基體表面,修飾后的復合材料通過原位熱解,轉變為中空納米管狀碳材料(圖3a)。二維層狀MOF材料熱解后可直接制備MOF衍生納米片,或剝離生成多層石墨烯納米帶(圖3b)。三維MOF衍生材料具有豐富的多級結構,如三維納米線列陣、碳納米管三維交聯結構、蜂窩狀納米結構等(圖3c)。不同維度的MOF衍生材料提供了金屬離子摻雜的多孔復合結構,在金屬鋰電池界面防護中均有應用,對其孔道結構、電子電導、離子電導、電催化性能的調控可進一步促進界面的傳質傳荷過程,緩解上述界面問題。Yi等55以SiO2納米球為模板,將鐵基MOF原位轉化成空心Fe3O4/碳復合材料,不僅通過配位聚合誘導的自組裝產生了微米級的空腔結構,還在MOF的外殼表面形成介孔,進一步提升鋰離子的傳導能力。Zhao等56使用鐵基MOF,通過熱解衍生出碳納米材料,具備良好的電催化活性,是貴金屬催化劑的替代材料之一。
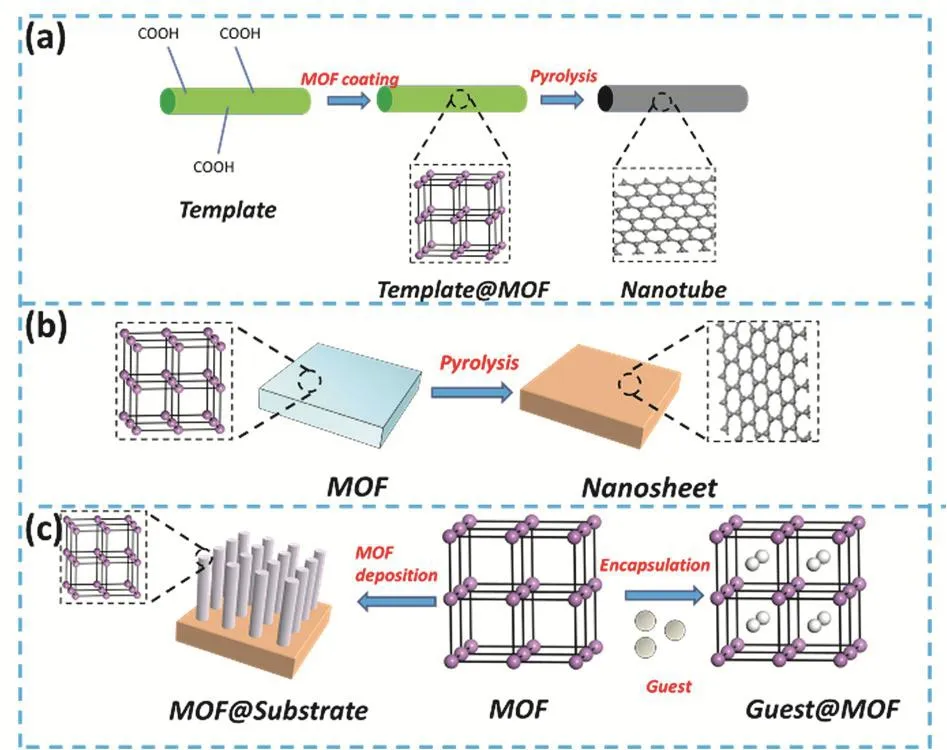
圖3 (a)一維,(b)二維,(c)三維MOF衍生物的納米結構合成示意圖54Fig. 3 Synthesis schematic of (a) one-dimensional,(b) two-dimensional, (c) three-dimensional MOF-derived materials nanostructure 54.
3 金屬鋰界面防護
金屬鋰具有高比容量(3860 mAh·g-1)、低還原電位(?3.04 VvsSHE)和較高的反應活性,是構建金屬鋰電池的必備電極材料。鋰金屬在實際的電化學循環過程中問題顯著。枝晶的生長容易刺穿隔膜,造成電池內短路,觸發熱失控等安全問題;界面失穩現象抑制了其電化學容量的發揮,造成了電化學性能的快速衰減。如何抑制鋰枝晶生長、死鋰生成、體積膨脹、庫倫效率低等問題是金屬鋰界面研究的重要方向。目前,大部分防護研究集中于SEI的均一化、穩定化,從界面傳質傳荷動力學調控金屬鋰的電沉積方式57,58。MOF材料具備良好的物理化學穩定性,其獨特的多孔結構、配位金屬的親鋰性為鋰離子的界面擴散提供了離子流均一化通道,在金屬鋰負極的界面保護上得到了廣泛應用。
3.1 鋰枝晶的生長機理
金屬鋰界面上鋰枝晶的生長問題始終是鋰金屬二次電池發展道路上的絆腳石,鋰枝晶的生長是多種因素共同作用下的結果,這些因素影響著鋰枝晶的生長速率與形貌。研究人員分別從溫度、過電位、電流密度、內應力等角度探討了鋰枝晶生長的過程。(1)在溫度控制的枝晶生長模型中,溫度一方面影響了鋰離子的傳輸行為,高溫可以促進鋰離子的擴散并間接改變鋰枝晶的形貌,另一方面溫度的變化可影響鋰金屬的力學性能。Aryanfar等59提出了熱松弛效應,該效應認為高溫促進了鋰離子的運動并使得鋰離子更易從金屬表面的凸起部分擴散到平坦部分;其二,Xu等60發現微米級別的鋰在室溫下屈服強度高達105 MPa,而在90 °C時屈服強度僅為35 MPa,產生了顯著的降低。(2)過電位對于鋰枝晶形貌影響的根源可能在于新生成的鋰的尺寸不同,Pei等61利用掃描電子顯微鏡(SEM)研究了金屬鋰在銅基體上的成核過程,如圖4a,b所示,隨著電流密度的增加鋰形核尺寸逐漸降低,鋰原子核的大小與過電位的倒數成正比。Monroe等62通過理論計算發現不同電流密度下鋰枝晶生長行為存在差異。在鋰枝晶生長早期,鋰枝晶的尺寸隨時間近似呈線性增長,而一段時間之后鋰枝晶生長加速。降低電流密度則有可能延長線性生長的時間并降低鋰枝晶生長的速度。(3) Kushima等63更為深入的研究了枝晶的生長過程,他們采用高倍率透射電子顯微鏡(TEM)觀察鋰枝晶的形核和生長,將鋰枝晶的生長過程分成了四個不同的階段。第一階段中球形鋰原子核在表面出現,其直徑與時間的平方根成正比;第二階段中,鋰枝晶開始生長并推動第一階段中生成的鋰原子核遠離表面,此時枝晶寬度不變而長度迅速增加;第三階段中枝晶生長速率顯著降低;第四階段中枝晶上形成了扭結,部分枝晶不再生長,并且伴隨著新枝晶的快速生長,扭結形成。(4) Xu等64提出了應力生長模型,他們發現在鋰金屬的電化學沉積過程中電極表面會產生應力,這種應力是鋰枝晶生長的驅動力。如圖4c所示,這種驅動力是在非平衡生長條件下,表面原子過度插入晶界引起的,鋰金屬中局部的蠕變為鋰枝晶的擠出提供了驅動力。
圖4 (a)不同容量下電流密度與形核尺寸的關系61;(b)過電位與形核尺寸的關系圖61;(c)應力驅動鋰枝晶生長的示意圖64Fig. 4 (a) Relationship between current density and nucleation size under different capacities 61, (b) relationship between overpotential and nucleation size 61, (c) schematic diagram of stress-driven lithium dendrite growth 64.
3.2 MOF基金屬鋰沉積骨架
MOF基金屬鋰沉積骨架是通過MOF材料在傳統碳基電極表面的表面修飾,或通過煅燒法制備出金屬/碳多維骨架結構,達到強化傳統碳材料的界面調控性能的目的。骨架的親鋰性位點與高比表面積能協同控制金屬鋰沉積的局部電流和均一度,實現高電流密度下鋰沉積的穩定性。Wang等65將由Zn(NO3)2·6H2O和2-甲基咪唑形成的ZIF-8材料作為前驅體,采用聚酰亞胺作為粘結劑,制備了ZnO/C電極,之后直接將熔融鋰注入ZnO/C電極制備了ZnO/C/Li電極,制備過程如圖5a所示。受益于ZnO/C電極界面上的羰基、含氮基團和氧化鋅等豐富的親鋰位點,鋰金屬可以順利注入。ZnO/C/Li電極有效降低了其中金屬鋰體積變化并抑制了鋰枝晶的生長。ZIF-8衍生材料修飾的金屬鋰界面,可以在10 mA·cm-2高電流密度下,穩定循環200周。Lyu等66利用2-甲基咪唑(C4H6N2)與Zn(NO3)2·6H2O反應制備了Zn-MOF,并以Zn-MOF作為前驅體采用3D打印技術制備了3DP-Zn-MOF材料,后在氮氣氛圍中煅燒制備了三維打印的氮摻雜碳骨架結構(3D-printed N-doped carbon framework,3DPNC)。3DP-NC具有分層孔隙結構,比表面積高達869 m2·g-1。在10 mA·cm-2的高電流密度,該骨架的鋰沉積容量可達到30 mAh·cm-2,同時具備小電流的長循環穩定性(1 mA·cm-2時穩定循環2000 h)和高庫倫效率(97.9%)。
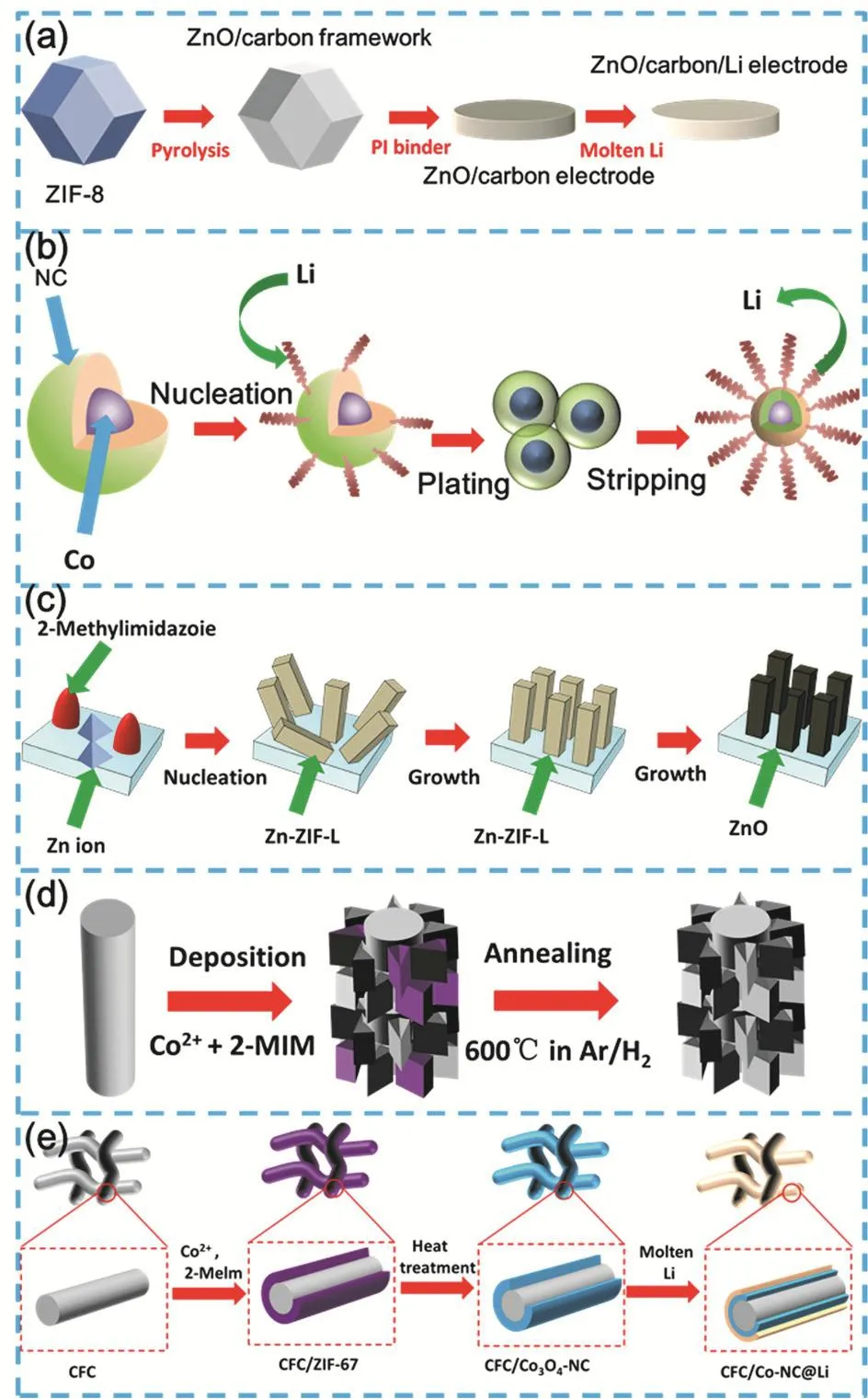
圖5 (a) ZnO/C/Li復合電極的制備流程示意圖65比例尺均為1 cm;(b)氮摻雜多孔石墨烯電極(Co@N-G)電鍍/剝離過程中的形態演變67;(c) LZNF合成示意圖68;(d)在碳布基底上生長MOF衍生的CN-Co納米片陣列的示意圖69;(e) CFC/Co-NC@Li電極的制作示意圖70Fig. 5 (a) Preparation process of ZnO/C/Li composite electrode 65, (b) morphological evolution during the electroplating/stripping process of nitrogen-doped porous graphene electrode (Co@N-G) 67, (c) schematic diagram of LZNF synthesis 68, (d) schematic diagram of growing MOF-derived CN-Co nanosheet arrays on the carbon cloth substrate 69, (e) schematic diagram of making CFC/Co-NC@Li electrode 70.
為了進一步改善沉積骨架的防護性能,MOF材料可與介孔碳、石墨烯、碳布等碳材料組成復合沉積骨架,加強材料的電子電導率和比表面積。Wang等67通過高溫碳化ZIF-67,制備鈷/氮雙摻雜多孔石墨烯電極(Co@N-graphene)。針對沉積/剝離過程中Co@N-graphene電極的形態演變研究表明(圖5b),碳化后的菱形十二面體結構有更好的結構穩定性以及高表面積,提供足夠的內/外部空間,穩定金屬鋰電極的沉積/剝離行為,從而抑制金屬鋰體積膨脹。MOF衍生物涂層中引入的Co、N等親鋰性雜原子,有效降低了鋰成核過電位和電極極化,使得在高電流密度下(15 mA·cm-2),80周循環后的金屬鋰電池仍保持較高的庫倫效率(90.4%)。類似的,Zhao等68將鋅基MOF材料生長在泡沫鎳材料表面,通過熱解制備出垂直排列的ZnO納米片的MOF衍生物材料(leaflike ZnO nanosheets on Ni foam,LZNF) (圖5c)。這種親鋰多孔骨架同樣能起到降低界面電流密度,誘導無鋰枝晶沉積,緩解體積膨脹的作用。在LZNF/Li電池庫侖效率較高(在1 mA·cm-2的150個周期中為98.5%),對稱電池(LZNF@Li/LZNF@Li)在5 mA·cm-2的電流密度下,可穩定循環250個周期。與金屬襯底相比,碳布具有較輕的密度、較穩定的化學性質、較大的表面積、豐富的孔隙結構和良好的電子導電性,同樣被廣泛用作MOF材料的生長基體和衍生物碳化基材。Zhou等69在三維碳布上生長Co基MOF材料,熱解后構建了修飾微小Co納米顆粒的氮摻雜碳納米片陣列(CC@CN-Co),并以此調節鋰金屬工作陽極中鋰的均質成核/生長行為(圖5d)。通過Li與雜原子的相互作用,N摻雜被證實具有引導Li均勻成核的作用。同時,良好的陣列結構可以增加活性表面積,促進鋰離子的均勻分布,從而解決鋰枝晶生長失控的問題。Jiang等70使用商品碳纖維布(CFC),將ZIF-67納米薄片原位生長和轉化形成了Co3O4嵌套和氮摻雜的多孔碳納米薄片陣列(Co3O4-NC)(圖5e),并將其用于鋰金屬復合負極的熔融鋰儲存。由于Co3O4與熔融鋰的化學反應性、氮摻雜物的鋰離子性質以及多孔結構,多孔Co3O4-NC納米薄片陣列可以顯著改變碳纖維布的弱Li潤濕行為,保證熔融鋰的快速注入。此外,的可濕性納米薄片陣列框架CFC/Co3O4-NC有良好的熔融鋰保持率,提供均勻的物理約束和快速沉積電荷轉移的氧化還原反應。特別是具有高比表面積和機械強度的CFC/Co3O4-NC基體可以降低局部電流密度,緩沖體積的變化,從而抑制循環過程中鋰枝晶的生長。
3.3 人造SEI膜
MOF基復合人造SEI膜為金屬鋰界面提供了有效的物理屏障,又通過化學成分修飾,增強了界面膜的離子通透性,能有效改善金屬鋰界面傳質傳荷過程。例如:傳統有機聚合物修飾層離子遷移數低(通常低于0.4),PEO基有機聚合物的鋰離子遷移數為0.2。通過MOF/有機聚合物的復合,同時合理設計MOF官能團,可顯著增大鋰離子遷移數,達到協同增強界面修飾層的離子傳輸和力學性能的效果。增加Fan等71通過如圖6a所示的流程制備出PVA與鋅基MOF的復合界面膜,圖6b示意了人造SEI膜對電極的保護過程。有機-無機復合人造SEI膜兼備高鋰離子導電性和良好的柔韌性,既均勻了鋰離子通量,抑制鋰枝晶的生成,又能有效的適應電極的體積變化,提高電池循環壽命。在3 mA·cm-2的高電流密度下仍能夠在250周循環后保持97.7%的庫倫效率。Qian等72的研究發現使用MOF-199制備的保護層同樣可以有效保護金屬鋰負極界面,提高電化學性能。圖6c展示了裸銅電極和保護后電極表面的金屬鋰沉積和剝離示意圖。依靠MOF致密、堅固的特性,從物理上抑制了鋰枝晶的生長,涂覆MOF-199的銅電極在1 mA·cm-2的電流密度下,350周循環后仍能夠保持97%以上的庫倫效率。MOF材料作為人造SEI膜時,其良好的離子通透性對鋰離子流的分布效果顯著。Shi等73提出了利用選擇性透過的方法來改善鋰離子的界面傳輸行為。如圖6d所示,生物中的隧道狀膜蛋白的隧道內部含有大量帶負電荷的官能團,其可以選擇性的允許特定陽離子通過(如Na+和K+)通過,同時靜電排斥陰離子(如Cl?)。如圖6e所示,他們采用UiO-66-NH2(UN)作為仿生離子通道,后將配體中的氨基通過化學修飾與SO3?基團連接并與鋰離子進行離子交換(記為UN-SLi),最后將UN-SLi與聚偏氟乙烯-聚氟丙烯(P(VdF-HFP))復合制成復合膜(UN-SLi-EM)。恒電位法測量后發現,采用UN-SLi-EM膜的對稱電池鋰離子遷移數(tLi+= 0.74)和鋰離子電導率(0.67 mS·cm?1)顯著提高。
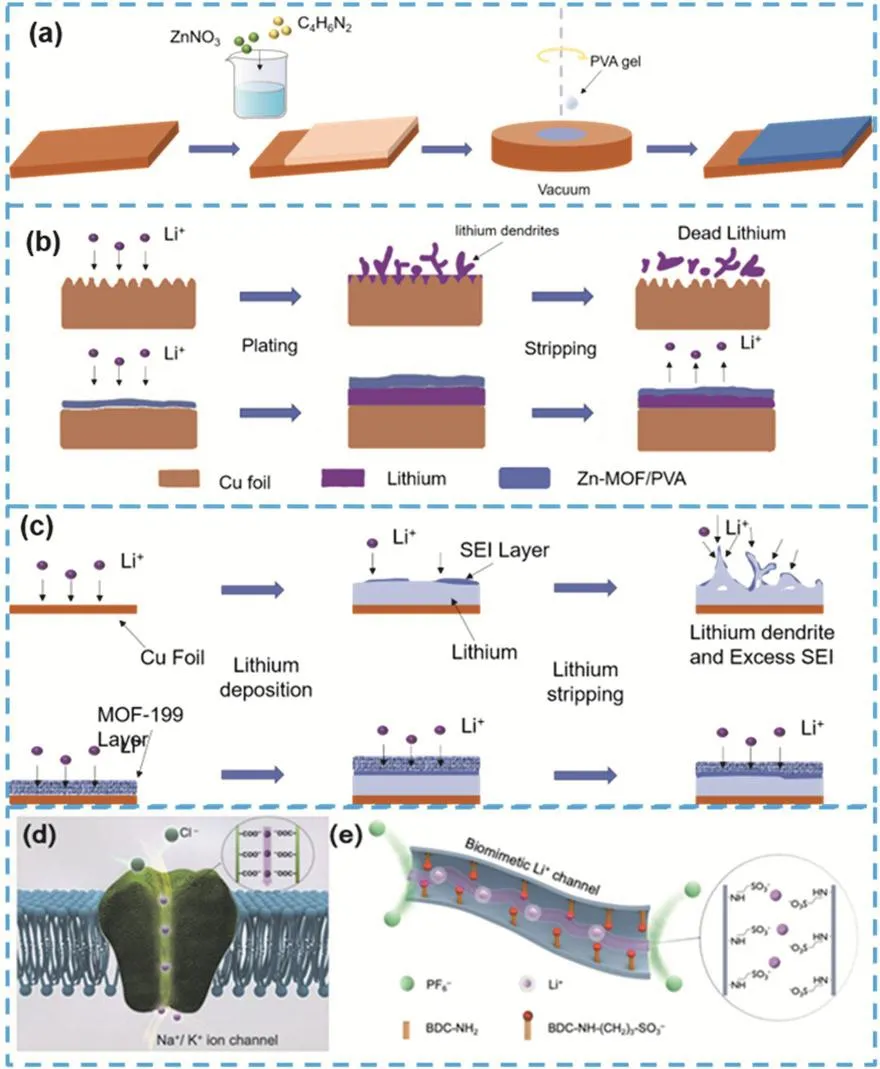
圖6 (a)制備PVA與MOF的復合界面膜的示意圖71;(b)裸銅箔上的鍍鋰/剝離行為,以及復合SEI膜對銅箔的保護圖71;(c)銅電極和具有MOF-199涂層的銅電極在循環過程中表面結構示意圖72;(d)細胞膜上帶有負電荷官能團的離子通道的示意圖73;(e) UN-SLi仿生離子通道的示意圖73Fig. 6 (a) Schematic diagram of preparing a composite interface film of PVA and MOF 71, (b) schematic diagram of copper foil protected by composite SEI film 71, (c) schematic diagram of the surface structure of copper electrodes and copper electrodes with MOF-199 coating during cycling 72,(d) schematic diagram of the biomimetic Li+ channel constructed by grafting anionic sulfonate groups on the pore channels of UN 73, (e) Schematic diagram of the UNSLi bionic ion channel 73.
3.4 電解液/電解質添加劑
無論是液態金屬鋰電池或固態金屬鋰電池體系,金屬鋰的界面不穩定性是其發展面臨的共性問題。在這方面,MOF作為電解液或電解質的功能性添加劑,在液態、固態電池體系均有較好的應用,被證明可抑制金屬鋰的失穩現象74。
在液態電池體系中,Chu等75發現將MOF添加于碳酸乙烯酯(EC)和碳酸二甲酯(DMC)的混合溶劑時(體積比為1 : 1),能有效穩定金屬鋰的電鍍/剝離過程,并抑制金屬鋰負極枝晶生長。通過循環性能的測試(圖7a-c),發現鋯基MOF添加劑(UiO-66)的電化學性能優于其他典型的銅基和鋁基MOF,在200周循環時,仍能夠保持大于95%的庫倫效率。其中MOF添加劑的結構穩定性、高孔隙率和電化學穩定性是穩定鋰負極的關鍵。在界面成膜過程中,UiO-66添加劑能提高了SEI中LiF的濃度,減少副反應的發生,均勻離子流分布,抑制了鋰沉積的擠壓變形以及斷裂。
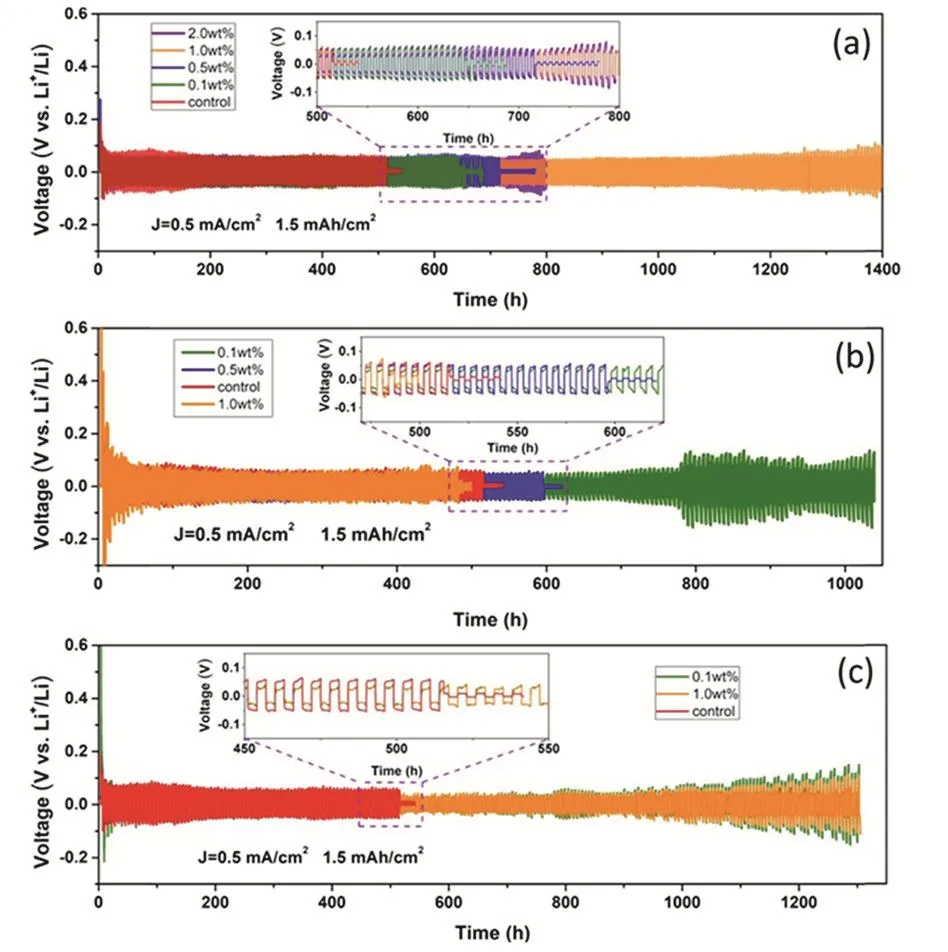
圖7 在面積容量為1.5 mAh·cm?2、電流密度為0.5 mA·cm?2的條件下,在電解質LiPF6-EC-DMC中添加不同比例(a) UiO-66、(b) HKUST-1和(c) MIL-101-NH2的Li|Li對稱電池的循環性能對比圖75;Fig. 7 Cycle performance of Li|Li symmetric battery with different ratios of (a) UiO-66, (b) HKUST-1 and(c) MIL-101-NH2 added to LiPF6-EC-DMC 75 under the condition of 1.5 mAh·cm?2 and 0.5 mA·cm?2.
MOF功能性添加劑應用于聚合物電解質體系時,其孔隙設計決定了鋰離子的傳輸性能;此外,MOF的多孔結構為電解液的吸附提供了充足的空間,有效提高復合聚合物電解質的保液性、熱穩定性和安全性76。Angulakshmi等77將含有胺基功能化的鋯基MOF@SiO2作為鋰硫電池的復合聚合物電解質(CPEs)的添加劑,由于MOF固有的多孔結構,形成了更均勻的離子傳導通道,不僅將CPEs提的離子電導率從7.6 × 10?7S·cm-2提高到8.1 × 10?6S·cm-2,而且具有更好的電極/電解質界面相容性,明顯的抑制了鋰枝晶的形成。Liu等78通過原位生成的方法制備了聚環氧乙烯(PEO)基的PEO/LiN(SO2CF3)2/MOF-5的聚合物電解質。由于MOF-5、N(SO2CF3)?2與PEO鏈中Lewis酸中心之間的相互作用,添加劑抑制了PEO的結晶性,使得離子電導率提高至 3.16 × 10?5S·cm-1。 使用PEO/LiN(SO2CF3)2/MOF-5電解質的LiFePO4半電池在0.2C、0.3C、0.5C、1C的倍率下放電容量分別為143、141、138和132 mAh·g?1(60 °C)。Stephan等79通過簡單的熱壓工藝制備了PEO/LiN(SO2CF3)2/Mg-BTC (BTC為三氯甲基碳酸酯)的復合聚合物電解質。單純的PEO/LiN(SO2CF3)2電解質分解溫度為190 °C左右,而Mg-BTC的添加使分解溫度提高至290 °C。復合聚合物電解質在20-70 °C溫度下的離子電導率處于10-8- 10-4S·cm?1,比單純的PEO/LiN(SO2CF3)2電解質高出2個數量級。此外,Mg-BTC的添加能有效降低鋰離子傳導的活化能(Ea,分別為1.3和0.6 keV)。Mg-BTC的Lewis酸中心和鋰離子相互作用能降低離子耦合,促進鹽的解離和有效傳導。采用PEO/LiN(SO2CF3)2/Mg-BTC的LiFePO4的半電池可提供了110 mAh·g-1的容量(70 °C,1C)。Zhu等80通過靜電吸附作用,將三氟甲基磺酰陰離子(Tf)錨定在鋯基UiO-66-NH2上,從而獲得優異的鋰離子導電電解質。其可調節的多孔結構為鋰離子輸送提供了獨特的通道。納米尺寸的MOF顆粒增加了電解質與電極界面的接觸,從而促進了離子的輸運動力學。該電解質高的離子電導率(25 °C時為2.07 ×10-4S·cm-1)、較高的鋰離子遷移數(0.84)均能夠為電池提供較好的電化學性能。在500圈充放電循環后,比容量可達到132 mAh·g-1,容量保持率為97.0%,同時在0 °C的低溫下,以1C倍率循環500周仍能保持91.1%的放電容量保持率。Zhang等81研究了一種基于金屬-有機骨架的獨特的納米孔結構固態電解質,該結構將含鋰的液態電解質(IEs)浸入到UiO-66框架中以獲得UiO/IEs。當這些填料加入到PEO基體中時,復合的電解質具較高的離子電導率(30 °C時為1.47 × 10?4S·cm?1)、5.2 V的電化學窗口以及0.47的鋰離子遷移數,與金屬鋰負極具有良好的相容性,從而有效抑制鋰枝晶生長,降低電解質與金屬鋰負極的界面電阻。在磷酸鐵鋰作為正極材料的全固態鋰金屬中電池的初始放電比容量為140.2 mAh·g-1,0.1C倍率下經過100個循環后仍能保持135.4 mAh·g-1的比容量。Wu等82用UiO-66吸收離子液體(Li-IL)并將其分散在PEO中制備出了PEO-n-UiO復合聚合物電解質,UiO-66的加入提高了電解質的離子導電性并抑制了PEO與電極的副反應。在30 °C時,含有40% UiO/Li-IL的PEO-n-UiO固態電解質的電導率增加到了1.3 ×10?4S·cm?1;同時在60 °C時,穩定沉積/剝離鋰的電流密度也提高到了500 μA·cm?2。在0.5C和60 °C的情況下,基于PEO-n-UiO固體電解質的電池初始放電容量約為151 mAh·g-1,并且循環100次之后容量保持率達到了95%。
4 MOF材料在隔膜界面保護中的應用
傳統隔膜與金屬鋰界面的不相容性是誘發金屬鋰界面離子流不均一的原因之一,隔膜的多孔特性加劇了鋰沉積的不均一性和枝晶的生長83。MOF基隔膜材料具備良好的離子通透性和OMs,能提高復合隔膜的離子傳導均一性、鋰親和性和離子電導率,協同穩定界面傳質傳荷過程84。
Zhang等46采用靜電紡絲技術制備了鋯基MOF UiO-66與PVA的復合隔膜材料。復合隔膜中的羥基和羧基發生酯化反應,形成三維交聯的復合膜(EMP)纖維網絡。電解液中的陰離子會與MOF顆粒的OMs發生絡合,從而提高鋰離子電導率與遷移數。表面形貌分析直接證實了改性隔膜對抑制枝晶生長的作用:改性隔膜能誘導鋰的緊湊、二維沉積,鋰金屬表面無明顯枝晶結構。這種隔膜/金屬鋰界面調控有助于降低極化,從而提高電池倍率和循環性能。
MOF材料對傳統隔膜的表面原位修飾是更具商業化前景的隔膜修飾和金屬鋰電池界面穩定策略。Bai等85制備了MOF@氧化石墨(MOF@GO)隔膜,在濾膜上原位生長MOF (HKUST-1)層后,過濾一定量的稀釋GO溶液,在均勻結晶的MOF納米顆粒上形成了平行的GO層,MOF與GO緊密結合,之后從濾膜上剝離MOF@GO隔膜,制備過程如圖8a所示。經N2吸附表征后HKUST-1的孔徑大約為0.9 nm,而在鋰硫電池中MOF@GO隔膜阻止了多硫化物向陽極的移動而對鋰離子的移動影響較小,因此采用MOF@GO隔膜的鋰硫電池初始放電容量達到了1126 mAh·g?1,并且在500圈仍可保持在800 mAh·g?1左右。針對商業化聚丙烯(PP)修飾使MOF復合隔膜研究更具產業化潛力。Zang等86在隔膜原位生長了導電金屬有機骨架Ni3(HITP)2(如圖8b),使得復合隔膜兼具優異的柔韌性和高比表面積(638.8 m2·g?1)。在鋰硫電池體系中,復合隔膜的導電側可抑制多硫化物穿梭,同時提高硫的利用率,絕緣側有助于避免正負極之間的接觸。采用Ni3(HITP)2/PP(聚丙烯)隔膜的鋰硫電池具有較高的容量、良好的倍率性能和循環穩定性,高載硫的電池(正極中硫載量8.0 mg·cm?2,硫質量含量70%)在200次循環后仍可提供7.24 mAh·cm?2的高面積容量。
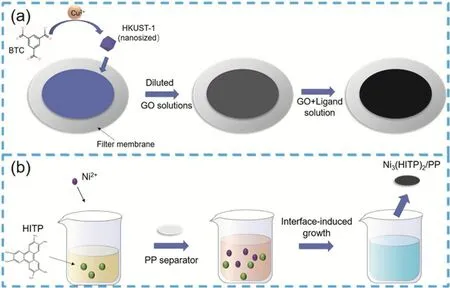
圖8 (a) MOF@GO隔膜的制備流程85;(b)制備Ni3(HITP)2/PP隔膜的示意圖86Fig. 8 (a) Preparation of MOF @ GO flow separator 85;(b) schematic diagram of preparing Ni3(HITP)2/PP separator 86.
除隔膜與電極的相互作用外,其與電解液的作用同樣值得重視。電解液中的痕量水分一直是制約電池安全性的因素,易引發陰極析氫反應(HER)與陽極析氧反應(OER),形成氣態產物導致電池內部壓力增大,同時消耗了大量的電解液87。Chang等88將Cu3(BTC)2材料(HKUST-1)作為隔膜同時兼具吸水劑的功能,HKUST-1隔膜抑制了電化學循環過程中陰極中過渡金屬的溶出,并且顯著抑制與痕量水引發的副反應產物(如:氟化鋰,碳酸乙烯酯的開環反應產物等)。當采用LiNi0.8Co0.1Mn0.1O2陰極時,在含水電解液中(含水量為200 × 10?6),400圈循環后,電池容量保持率高達72%。
5 MOF材料在正極界面保護中的應用
正極活性物質溶出是金屬鋰界面不穩定的誘因之一。正極溶出的過渡金屬離子、多硫化物等會在負極界面發生電還原沉積,從而打破金屬鋰界面的穩定性。此外,負極界面成分同樣會發生溶解、電化學遷移,參與正極界面膜的形成和生長,破壞正極固態電解質膜(CEI)的穩定性。因此,由于正負極界面的相互作用,金屬鋰界面穩定性也會受到正極界面穩定性的影響,在金屬鋰電池、鋰硫電池、鋰氧電池等高比能電池體系中表現出不同的界面失效特性。
5.1 過渡金屬化合物界面防護
過渡金屬基正極材料是設計高比能、高電壓金屬鋰電池的有力競爭者,例如:LiCoO2、LiNi1?x?yCoxMnyO2等。正極CEI膜決定過渡金屬離子的泄露和電解液的復分解89。Xie等90使用Zr基MOF (UiO-66-F4)作為前驅體,在Li1.2Mn0.54Co0.13Ni0.13O2這種富鋰層狀氧化物(LLO)上原位包覆MOF衍生ZrO2層(ZrO2@LLO)。圖9a-f通過SEM觀測到碳酸鹽前驅體(a,d)、原始LLO (b,e)和ZrO2@LLO的微觀形貌差異。由于原生納米顆粒之間存在大量的孔隙,增大了ZrO2@LLO微球的比表面積,從而增加了循環過程中的鋰離子擴散通道。同時氟離子的摻雜及其良好的親鋰性,降低了鋰離子的擴散阻力。這種元素摻雜可以有效地穩定晶體結構,抑制第一個周期中氧空位的形成,從而將首周庫倫效率從62%提高到72%,1C充放電倍率下循環200周后容量保持171.3 mAh·g?1。Jia等91將的ZIF-67粉末與碳酸鋰混合,高溫燒結鋰化,將ZIF-67納米多面體轉化為LiCoO2顆粒,得到了MOF衍生的碳包覆核-殼結構LiCoO2@C,根據密度泛函理論得到LiCoO2@C提供了明顯有效的鋰離子擴散路徑,改善了電化學導電性和碳與LiCoO2本體之間的強電子相互作用,有利于提高電化學性能。在700 °C下燒結得到的LiCoO2@C-700有著最好的性能,在1C充放電倍率下200個循環后,保持了171.1 mAh·g-1的容量。

圖9 (a,d)碳酸鹽前驅體;(b,e)原始LLO和(c,f) MDZ@LLO的SEM圖像圖90Fig. 9 SEM image of (a, d) carbonate precursor; (b, e) original LLO and (c, f) MDZ@LLO 90.
5.2 鋰硫電池體系MOF界面穩定策略
單質硫的高理論容量(1675 mAh·g-1)受到多硫化物穿梭效應的影響,難以獲得長循環穩定性92,93。多硫化物穿梭引起的金屬鋰負極失穩現象,也是鋰硫體系中金屬鋰負極失效的典型機理。憑借多孔結構、較大的孔隙率、高表面積和功能性吸附位點,MOF能捕獲電極/電解液界面處滲漏的多硫化物,一方面穩定金屬鋰界面的鋰離子傳輸過程,另一方面提高正極硫的利用率和穩定性94。Zhou等95認為在制備高性能MOF儲硫電極時,除了要考慮MOF材料的結構,還要考慮客體離子的尺寸、化學環境、電壓范圍等因素。由于擴散的特征時間常數t正比于擴散長度L的平方(t≈L2/D,D為離子擴散率),通過減小材料的尺寸可以明顯降低Li+/e?擴散時間。MOF衍生的過渡金屬氧化物/碳復合材料是一類典型的抑制多硫化物穿梭的吸附材料,其多孔形態既增加了多硫化物吸附位點,又能化解金屬鋰負極傳質不均一的問題。在Liu等96的研究中,設計并制備了Fe基MOF(MIL-53)衍生的Fe3O4@C的多孔八面體結構。當其用作鋰硫電池硫載體時,極性Fe3O4@C中心可通過化學相互作用捕獲多硫化物,進而加速多硫化物的轉化。原位形成的多孔碳層保留了MOF材料的形態,具有納米級孔隙排列,可以提供足夠的表面來吸收和限制多硫化物。Fe3O4@C的高電子電導率有利于加速電子傳遞,增強反應動力學,減少多硫化物的遷移,協同提高了Fe3O4@C的硫正極利用率,穩定了正極、負極界面和電池整體性能。MOF及其衍生物作為擔載硫的基底,也是抑制多硫化物穿梭的主要策略之一。Walle等97將ZIFs/CNF (碳納米纖維)作為反應物,在氮氣氛圍下加熱至900 °C得到了NC (氮摻雜立方碳)/CNT (碳納米管)復合材料。NC/CNT復合物具有高比表面積(288.8 m2·g-1)和小孔徑體積的特點(0.3 cm3·g-1),能夠負載超高量的硫(3.6 mg·cm-2)。合金化反應過程中,NC/CNT不僅可以吸附多硫化物,而且其獨特的孔徑結構可以實現多硫化物的限域作用,協同提高電子電導率。復合結構中的氮在材料中提供了活性位點,具有吸附可溶性多硫化物的能力。在0.5C的電流密度下120個循環后比容量保持在674.4 mAh·g-1。
Han等98通過將八面體結構的UiO-66復合到常規的PP隔膜上,通過N2吸附解吸分析得到MOF材料存在大量約為0.5-0.6 nm的微孔。該復合隔膜的MOF孔徑小于多硫化物的尺寸,利用離子篩效應,選擇性透過鋰離子而阻礙多硫化物的溶出、穿梭。在100次充放電循環后,裝載MOF改性隔膜的鋰硫電池中,金屬鋰負極中硫含量僅為4.58%,顯著低于未修飾的PP隔膜(硫含量高達30.69%),證明了MOF復合隔膜在抑制多硫化物穿梭方面的作用。
Xi等99提出了一種利用碳化MOF負載硫制備鋰硫電池正極結構的方法。將四種Zn基MOF材料經過氬氣氛圍900 °C煅燒后在155 °C下與硫反應制備出具有不同層次孔結構的含硫多孔碳正極材料。結果表明,具有較高介孔體積(2-50 nm)的MOF衍生碳制成的正極材料,其初始放電容量增加,而具有較高微孔(< 2 nm)體積的碳,其正極材料具有較好的循環穩定性。從而為設計具有分級孔徑分布的碳基硫正極提供指導意義。
MOF結構中的金屬位點對吸附多硫化物,減緩鋰硫電池穿梭效應也具有積極的作用。Li等100通過將Co-O4基團連接在2D的MOF納米薄片上,與細菌纖維素(BC)逐層組裝成為BC/2D MOF-Co的隔膜材料,Co-O4基團中O原子對鋰離子有強吸附性,從而使鋰離子通量均勻化,穩定Li的剝離和沉積。同時在MOF-Co納米片表面的Co單原子可以通過Lewis酸堿相互作用有效地固定多硫化物,限制多硫化物的擴散,提高硫的利用率。此外,通過調控MOF中的孔結構,能從根本上抑制多硫化物的溶解。
5.3 鋰氧電池界面協同調控
界面穩定性、催化劑的穩定性和活性同樣是鋰氧電池發揮其高比能量的關鍵101,102。金屬鋰負極和多孔碳/催化劑正極界面共同決定了鋰氧電池的界面穩定性。過氧化鋰在多孔碳層的堆積、金屬鋰界面的副反應,都造成了鋰氧電池的高電極極化、低循環穩定性的特性。傳統的貴金屬催化劑(鉑和銀103,以及二元、三元復合催化劑Pt-Cr104,Pt-Fe105,106,Pt-Cr-Cu107,Pt-Fe-Co108等)存在成本高、金屬資源短缺等問題,限制了貴金屬催化劑在商業化鋰氧電池中的應用。MOF的多孔結構以及金屬活性位點為鋰氧電池提供了高催化活性和界面穩定性,是貴金屬催化劑的理想替代者。Jiang等109以ZIF-67為前驅體模板,通過低溫退火處理(溫度低于350 °C時),得到了骨架最為完整的Co3O4納米籠狀結構的催化劑(圖10a)。該結構具有良好的電荷轉移能力以及O2和鋰離子的擴散能力,其擁有更多的氧空位,提供了更多的活性位點。在納米結構和活性位點的協同作用下,鋰氧電池的正負極界面更加穩定,裝配的鋰空電池能夠實現深度充放電的穩定性(15500 mAh·g-1),在0.5 A·g-1的充放電電流密度下,132周的循環后能夠保持在1000 mAh·g-1的放電容量。同樣是Co3O4,Gong等110采用液相沉積方法,在柔性碳紡織材料上直接生長了二維金屬有機骨架(ZIF-L)。以MOF作為模板,先后在500 °C的氬氣和350 °C空氣中進行熱處理,得到了生長在柔性碳纖維織物(CT)上的Co3O4納米薄片(圖10b),由于有機成分的分解而產生多孔結構,得到的Co3O4/CT復合材料具有高比表面積和分層孔隙結構,有利于鋰離子和O2的擴散,為正極的還原產物Li2O2提供了很大的空隙空間。網格化的Co3O4納米薄片和無粘結劑的設計,保證了催化劑內部快速的電子傳遞,降低了催化劑與碳材料之間的界面阻抗。該結構具有足夠的Li2O2/正極接觸界面、良好的雙功能催化性能和足夠的Li2O2調節能力,從而提高了鋰氧電池的電化學性能。這種無粘結劑的Co3O4/CT正極提供6509 mAh·g-1的高容量。Yuan等111采用超薄二維金屬有機骨架(2D MOF)同樣改善了Li-O2電池的性能,具有高的氧氣可及性、開放的催化活性位點和較大的表面積。通過超聲合成方法,合成并干燥制備了二維金屬氧化物2D Mn-MOF。因為2D Mn-MOF具有開放的催化位點,較大的比表面積,特別是高電催化活性的Mn-O官能團,有利于有效的電解質/電催化活性位點接觸和O2與鋰離子的傳質,從而在Li-O2電池中提供良好的界面催化效果,電化學過程如圖10c所示。在基于Mn-MOF的Li-O2電池獲得了約9500 mAh·g-1的容量。
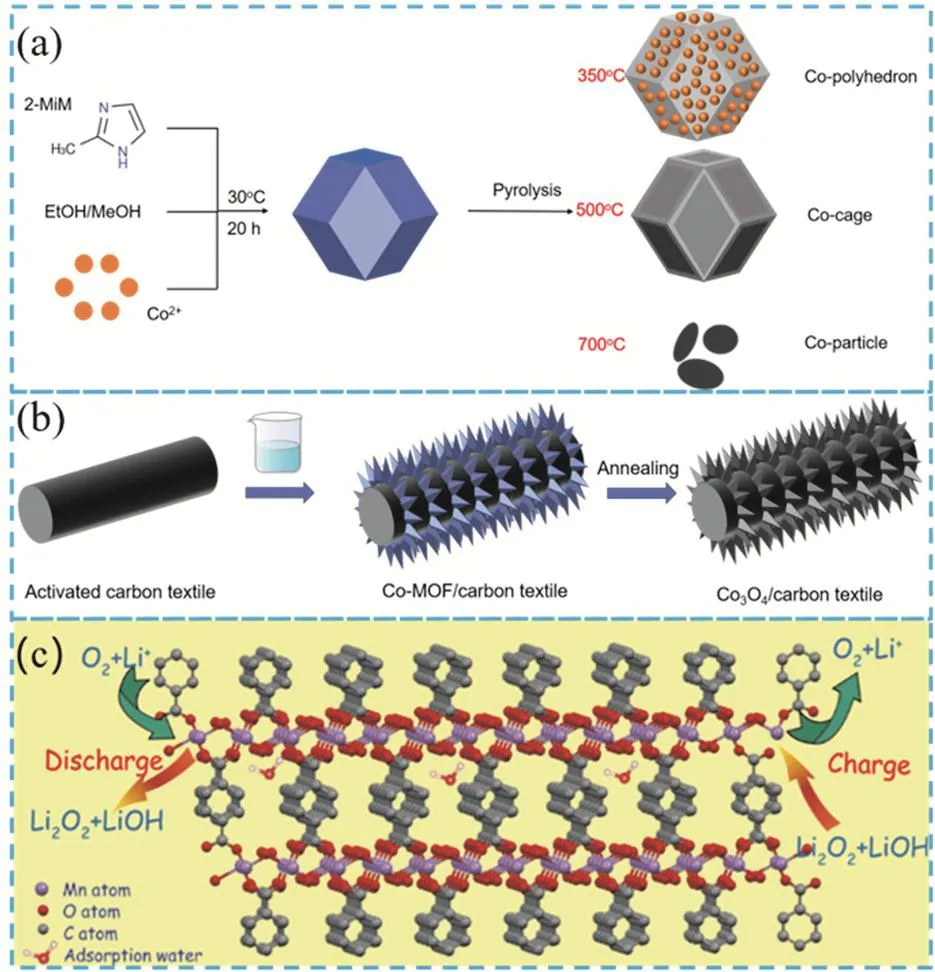
圖10 (a)以ZIF-67為模板合成三種Co納米結構材料的示意圖109;(b)在碳基底上生長Co3O4納米薄片的示意圖110;(c)二維Mn-MOF上可逆形成放電產物的電化學過程示意圖111Fig. 10 (a) Schematic diagram of the synthesis of three Co nanostructure materials using ZIF-67 as a template 109,(b) schematic diagram of growing Co3O4 nanosheets on a carbon substrate 110, (c) schematic diagram of the electrochemical process of reversibly forming discharge products on two-dimensional Mn-MOF 111.
6 總結和展望
高比能金屬鋰電池存在的典型界面問題,如電解液副反應、溶劑共嵌入、鋰枝晶生長和較大的界面阻抗等界面問題限制了其進一步發展,金屬鋰的高容量、高反應活性和不可控界面失穩現象仍是金屬鋰使用過程中的矛盾和突破難點,限制了金屬鋰電池的應用。從溫度、過電位、電流密度、內應力等角度,鋰枝晶的生長模型得到了深度理解。近年來,MOF及其衍生材料在解決高比能二次電池界面問題的研究得到了廣泛的研究和顯著的發展。基于金屬鋰界面的挑戰與問題,本文詳細的總結了MOF及其衍生物材料在金屬鋰電池體系中的應用,主要包括金屬鋰負極界面的改善策略,隔膜的修飾改性,及電解液、電解質的MOF添加劑的作用機理。通過對于MOF材料的配體以及金屬節點的合理選擇,可以實現孔徑的調整,其固有結構的多樣性帶來的高鋰離子電導率能夠為電池中各種界面處的離子交換提供幫助,穩定的骨架結構帶來的機械強度也能夠保護電極產生的體積變化,開放金屬位點以及功能性官能團能夠作為催化活性位點提供界面催化能力。
然而現有MOF研究多集中于源頭機理的可能性探索,在金屬鋰軟包和高容量電池體系中,金屬鋰電池界面將面臨更復雜的界面電化學過程,其界面面積及使用工況與實驗室模擬環境差異較大。因此,針對MOF的應用,更需要在實際的軟包電池體系中探討界面的保護作用。其次,MOF及其衍生物作為電極材料,在電池循環過程中仍會存在結構不穩定、反應物堵塞結構中空腔,以及固有導電性差等問題依然存在。導電性良好、大空腔、結構穩定的MOF材料依然需要繼續發展。特別是針對二次電池界面的MOF設計,仍有很大的改進空間。再次,MOF材料與不同電解液體系的適配性尚未解決。隨著高電壓正極材料和高壓電解液的發展,MOF材料修飾高壓正極材料時,電解液/MOF、正極固態電解質膜/MOF之間的穩定性、匹配性等仍需大量的基礎研究。最后,MOF材料在二次電池高低溫(特別是低溫環境)、快充快放、過充過放等極端條件下,對于二次金屬鋰電池界面的防護效果、枝晶抑制機制、防過充過放保護機制等,是MOF材料更值得深入探討和應用的方向。金屬鋰電池仍然處于應用研究的初期階段,界面問題仍是困擾金屬鋰電池發展的關鍵科學問題。合理設計MOF的金屬節點/有機配體、孔結構、無機/有機復合材料,深刻認識MOF材料在實際金屬鋰電池體系中的防護機理,將有助于推進金屬鋰電池的發展與實際應用。
猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34
當代陜西(2020年13期)2020-08-24 08:22:02
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
制造技術與機床(2017年5期)2018-01-19 02:49:17
金秋(2017年4期)2017-06-07 08:22:16
中國材料進展(2016年10期)2016-12-26 06:50:20
濰坊學院學報(2016年2期)2016-12-01 13:00:11
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
新聞傳播(2015年11期)2015-07-18 11:15:04
