親水作用液相色譜脫除人參提取物中農(nóng)藥殘留
2021-03-10 09:05:10孫伶俐郭秀潔吳立冬段正超王超然2王聯(lián)芝
色譜 2021年4期
孫伶俐, 劉 佳, 郭秀潔, 吳立冬, 段正超, 王超然2,*, 王聯(lián)芝*
(1.湖北民族大學(xué),湖北 恩施 445000;2.中國(guó)科學(xué)院大連化學(xué)物理研究所,遼寧 大連 116023;3.中科院大化所中國(guó)醫(yī)藥城生物醫(yī)藥創(chuàng)新研究院,江蘇 泰州 225300;4.中國(guó)水產(chǎn)科學(xué)研究院,北京 100141)
人參(PanaxginsengC.A.Mey)為五加科植物人參的干燥根和根莖,具有大補(bǔ)元?dú)?復(fù)脈固脫,補(bǔ)脾益肺,生津止渴,安神益智的功效[1],是我國(guó)著名的名貴中藥之一,被譽(yù)為“百草之王”[2,3]。人參提取物具有抗疲勞、抗衰老和保護(hù)心血管等諸多功效,被廣泛應(yīng)用于國(guó)內(nèi)外保健品和膳食補(bǔ)充劑中,21世紀(jì)以來(lái)一直是我國(guó)出口美國(guó)最暢銷的中藥提取物之一[4-7]。而隨著各個(gè)國(guó)家和地區(qū)對(duì)植物提取物中農(nóng)藥殘留的監(jiān)管日趨嚴(yán)格,農(nóng)藥殘留超標(biāo)問(wèn)題已成為制約我國(guó)植物提取行業(yè)發(fā)展的重要因素,其中尤以人參提取物中的農(nóng)殘備受關(guān)注[8-12]。如美國(guó)食品藥品管理局(food and drug administration,FDA)對(duì)人參提取物中農(nóng)殘檢測(cè)多達(dá)500多項(xiàng),且限量要求多為0.001~0.01 mg/kg,歐洲和日本雖然限量要求相對(duì)較為寬松,但檢測(cè)項(xiàng)目也分別達(dá)到了400多項(xiàng)和200多項(xiàng),極大地限制了我國(guó)人參提取物的出口[8]。
目前,常用的人參及人參制品中農(nóng)藥殘留的脫除方法有有機(jī)溶劑法[13,14]、大孔樹脂吸附法[15-17]和超臨界萃取法[18-21]等。李廣濤等[22]使用有機(jī)溶劑萃取法將人參提取物中腐霉利含量由44.51 mg/kg降至2.47 mg/kg,人參皂苷回收率為82.83%。該方法對(duì)腐霉利有一定的脫除效果,但未徹底脫除且人參皂苷損失嚴(yán)重;趙麗娟等[23]利用液液萃取和大孔吸附樹脂法有效脫除人參莖葉中多種農(nóng)藥殘留,但需使用氯仿、石油醚等有機(jī)溶劑,易對(duì)樣品和環(huán)境造成污染;韓玉謙等[24]利用超臨界1,1,1,2-四氟乙烷萃取技術(shù)可有效脫除人參中的有機(jī)氯農(nóng)殘,但易對(duì)樣品造成二次污染且設(shè)備運(yùn)行成本較高。
Alpert[25]在1990年首次提出親水作用液相色譜(HILIC)的概念。HILIC主要采用極性固定相,水和有機(jī)溶劑為流動(dòng)相,因其具有流動(dòng)相組成簡(jiǎn)單、分離效率較高且與質(zhì)譜有良好的兼容性等優(yōu)勢(shì)而越來(lái)越受到關(guān)注和重視,特別適用于分離強(qiáng)極性、帶電荷的親水化合物[26,27]。本實(shí)驗(yàn)根據(jù)農(nóng)殘和人參皂苷的結(jié)構(gòu)特點(diǎn),利用兩類化合物間親疏水性和相對(duì)分子質(zhì)量的差異,在HILIC柱上分離人參皂苷和農(nóng)殘兩類化合物,解決了現(xiàn)有脫除方法脫除不徹底、適用農(nóng)殘種類少、人參總皂苷損失率高和可能被二次污染等問(wèn)題,并且安全高效、能較大程度地提高人參總皂苷純度。
1 實(shí)驗(yàn)部分
1.1 儀器與試劑
Waters e2695型高效液相色譜儀(美國(guó)Waters公司),DAC50-動(dòng)態(tài)軸向壓縮柱(江蘇漢邦科技有限公司),TSQ 8000 GC-MS/MS(美國(guó)Thermo Fisher Scientific公司),Milli-Q Integral 115超純水系統(tǒng)(美國(guó)Millipore公司)
乙醇(食品級(jí),泰州蘇北化學(xué)試劑有限公司),乙酸乙酯、乙腈(質(zhì)譜純,德國(guó)默克公司),乙酸(分析純,國(guó)藥集團(tuán)化學(xué)試劑有限公司),石墨化炭黑(艾覽化工科技有限公司),十八烷基硅烷鍵合硅膠(C18)、N-丙基乙二胺(PSA)、硅膠、30 μm-C18YE、60 μm-Click XIon(華譜科儀科技有限公司),人參提取物(純度 HPLC≥59.8%)、嘧菌酯(純度≥99%)購(gòu)自上海源葉生物科技有限公司,Rg1、Re、Rf、Rb1、Rc、Rb2、Rd純度均≥98%,購(gòu)自成都植標(biāo)化純生物技術(shù)有限公司,五氯硝基苯(純度≥94%)、五氯苯胺(純度≥96%)、丙環(huán)唑(純度≥97%)、多菌靈(純度≥97%)、異菌脲(純度≥98%)、腐霉利(純度≥98%)均購(gòu)自阿拉丁試劑有限公司,113種農(nóng)藥殘留混合對(duì)照品購(gòu)自天津阿爾塔科技有限公司。
1.2 儀器條件
色譜保留行為評(píng)價(jià)反相色譜 C18YE色譜柱(250 mm×4.6 mm,10 μm);流動(dòng)相為乙醇(A)和純水(B)。洗脫程序?yàn)?~12 min,0%A;12~13 min,0%A~60%A;13~28 min,60%A;28~29 min,60%A~90%A;29~40 min,90%A。柱溫為30 ℃;檢測(cè)波長(zhǎng)為203 nm。
色譜保留行為評(píng)價(jià)親水色譜 Click XIon色譜柱(250 mm×4.6 mm,5 μm);流動(dòng)相為乙醇(A)和純水(B)。洗脫程序?yàn)?~12 min,95%A;12~13 min,95%A~60%A;13~21 min,60%A;21~22 min,60%A~40%A;22~28 min,40%A。流速為1.0 mL/min;柱溫為30 ℃;檢測(cè)波長(zhǎng)為203 nm。
皂苷樣品液相色譜分析 Click XIon色譜柱(250 mm×4.6 mm,5 μm);流動(dòng)相為乙腈(A)和純水(B)。洗脫程序?yàn)?~20 min,90%A~60%A。流速為1.0 mL/min;柱溫為30 ℃;進(jìn)樣量為10 μL;檢測(cè)波長(zhǎng)為203 nm。
親水色譜制備 DAC50-動(dòng)態(tài)軸向壓縮柱,UV檢測(cè)器,檢測(cè)波長(zhǎng)為203 nm,Click XIon色譜填料(填料重量300 g,粒徑60 μm),流速60 mL/min,以95%乙醇溶液為溶劑,將30 g人參提取物樣品溶解為600 mL,采用泵上樣方式,以乙醇(A)和純水(B)作為流動(dòng)相,洗脫程序?yàn)?~10 min,上樣;10~20 min,95%A淋洗;20~30 min,90%A洗脫得到溶液Ⅰ;30~45 min,60%A洗脫得到溶液Ⅱ,45~55 min,40%A洗脫得到溶液Ⅲ。
氣相色譜 色譜柱為Thermo農(nóng)殘專用柱(30 m×0.35 mm,0.25 μm);載氣為高純氦氣,流速1.2 mL/min;進(jìn)樣口溫度270 ℃;色譜柱升溫程序?yàn)?40 ℃保持1.5 min,以25 ℃/min升至90 ℃,保持1.5 min,再以25 ℃/min升至180 ℃,保持1.5 min,以5 ℃/min升至280 ℃,最后以10 ℃/min升溫到300 ℃,保持5 min;進(jìn)樣方式為不分流進(jìn)樣;進(jìn)樣量為1 μL。
質(zhì)譜 電子轟擊源70 eV,離子源溫度300 ℃,傳輸線溫度280 ℃;數(shù)據(jù)采集方式為多反應(yīng)監(jiān)測(cè)(MRM)模式。
1.3 標(biāo)準(zhǔn)溶液的配制
準(zhǔn)確吸取1 mL質(zhì)量濃度為10 mg/L的113種農(nóng)藥殘留混合對(duì)照品于10 mL容量瓶中,加入乙腈定容,得到質(zhì)量濃度為1 mg/L的混合標(biāo)準(zhǔn)儲(chǔ)備溶液。使用乙腈稀釋對(duì)照品溶液,配制成質(zhì)量濃度為5、20、100、300、600、1 000 μg/L的混合對(duì)照品溶液,儲(chǔ)存于-18 ℃?zhèn)溆谩?/p>
1.4 供試品溶液的配制
工藝條件優(yōu)化中的供試品溶液 精密稱定人參提取物0.250 g 3份,分別置于5 mL、10 mL、10 mL 3個(gè)容量瓶中,在1個(gè)5 mL和1個(gè)10 mL的容量瓶中,分別加入五氯硝基苯、五氯苯胺、腐霉利各1 mg,然后加入95%乙醇充分溶解,定容,搖勻,得到未添加農(nóng)藥標(biāo)樣的25 g/L人參提取物供試品溶液,添加農(nóng)藥標(biāo)樣的25 g/L人參提取物供試品溶液和添加農(nóng)藥標(biāo)樣的50 g/L人參提取物供試品溶液,分別標(biāo)記為原液Ⅰ、Ⅱ、Ⅲ。
農(nóng)藥殘留檢測(cè)中的供試品溶液 參照2015版《中國(guó)藥典》第四部多種農(nóng)藥殘留量的測(cè)定中供試品溶液的制備方法制備[28]。
1.5 定量及純度計(jì)算方法
HPLC分析含農(nóng)殘人參提取物試樣時(shí),農(nóng)殘脫除率和人參總皂苷(以Rg1、Re、Rf、Rb1、Rc、Rb2、Rd 7種皂苷計(jì)算,下同)回收率計(jì)算公式如下:
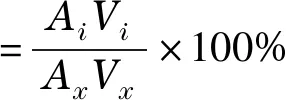
(1)
式中:Ai為所分析餾分中人參皂苷(農(nóng)殘)的總峰面積,Vi為所分析餾分的體積;Ax為原液分析時(shí)農(nóng)殘或人參皂苷的總峰面積,Vx為原液的上樣體積。
根據(jù)體積和峰面積計(jì)算人參總皂苷樣品純度,公式如下:
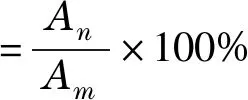
(2)
式中:An為所分析樣品溶液中人參皂苷的總峰面積;Am為所分析樣品溶液中所有成分的總峰面積。
根據(jù)上樣時(shí)原液中人參提取物的質(zhì)量濃度、上樣體積及色譜柱中填料質(zhì)量計(jì)算上樣量,公式如下:
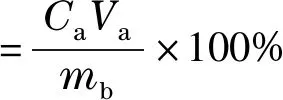
(3)
式中:Ca為原液中人參提取物的質(zhì)量濃度;Va為原液的上樣體積;mb為色譜柱中填料質(zhì)量。
2 結(jié)果與討論
2.1 農(nóng)藥與人參總皂苷色譜保留行為評(píng)價(jià)
基于疏水保留原理的大孔樹脂是目前工業(yè)上用于植物提取物精制,提高有效成分含量的主要工藝手段[29,30]。在樹脂精制過(guò)程中也可以同時(shí)去除大量的農(nóng)藥殘留,但仍有部分農(nóng)藥會(huì)與有效成分共流出,并在精制過(guò)程中一起被富集,從而在最終的產(chǎn)品中嚴(yán)重超標(biāo)。本實(shí)驗(yàn)以人參提取物中易超標(biāo)的7種農(nóng)藥標(biāo)準(zhǔn)品和市售總皂苷樣品為例,對(duì)比了其在反相色譜填料和親水色譜填料上的保留行為差異。為適應(yīng)后續(xù)工業(yè)生產(chǎn)工藝的開發(fā),模擬制備液相色譜方法,采用不同比例的乙醇水體系進(jìn)行等度洗脫。結(jié)果如圖1所示:在反相色譜中,除五氯硝基苯和五氯苯胺保留較強(qiáng),能與皂苷類成分顯著分離外,多菌靈、嘧菌酯、腐霉利、異菌脲和丙環(huán)唑5種農(nóng)藥都與人參總皂苷的保留相近;而在親水色譜中,與總皂苷成分保留情況不同,7種農(nóng)藥在95%乙醇條件下僅有微弱保留,與總皂苷樣品實(shí)現(xiàn)了良好的分離。Click XIon固定相典型的表面化學(xué)結(jié)構(gòu)和典型人參皂苷及7種農(nóng)藥的結(jié)構(gòu)式如圖2所示。從農(nóng)藥與總皂苷成分在反相色譜柱上的保留規(guī)律來(lái)看,基于類似的疏水保留機(jī)理,樹脂純化后的總皂苷樣品,即使采用分離柱效更高的球形硅膠基質(zhì)的反相填料也很難將農(nóng)殘進(jìn)一步脫除。而在Click XIon親水色譜中,由于固定相表面同時(shí)具有帶正負(fù)電荷的官能團(tuán),使其具有較強(qiáng)的親水性[27]。根據(jù)之前文獻(xiàn)報(bào)道,皂苷糖基上的羥基可與Click XIon固定相表面的羧基形成氫鍵,而這種氫鍵作用是皂苷在Click XIon柱上保留的主要次級(jí)作用,使得總皂苷樣品因?yàn)樘擎湹拇嬖诙哂休^強(qiáng)的親水保留[31],而這7種農(nóng)藥由于相對(duì)分子質(zhì)量較小,親水性較差,在親水色譜填料上保留微弱。正是基于兩者保留行為上的巨大差異,本實(shí)驗(yàn)擬開發(fā)一種適用于人參提取物中農(nóng)殘脫除的方法。
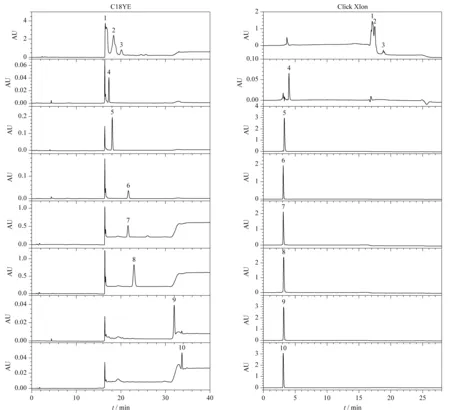
圖 1 7種農(nóng)藥與人參總皂苷標(biāo)準(zhǔn)品在反相和親水色譜柱上的色譜圖Fig.1 Chromatograms of seven pesticide residues and total ginsenosides on RPLC and HILIC columnsMobile phase A:ethanol;mobile phase B:water.Elution program for C18YE (250 mm×4.6 mm,10 μm):0-12 min,0%A;12-13 min,0%A-60%A;13-28 min,60%A;28-29 min,60%A-90%A;29-40 min,90%A.Elution program for Click XIon (250 mm×4.6 mm,5 μm):0-12 min,95%A;12-13 min,95%A-60%A;13-21 min,60%A;21-22 min,60%A-40%A;22-28 min,40%A.Peak identifications:1,2,3.ginsenosides;4.carbendazim;5.azoxystrobin;6.procymidone;7.iprodione;8.propiconazole;9.pentachloroaniline;10.quintozene.
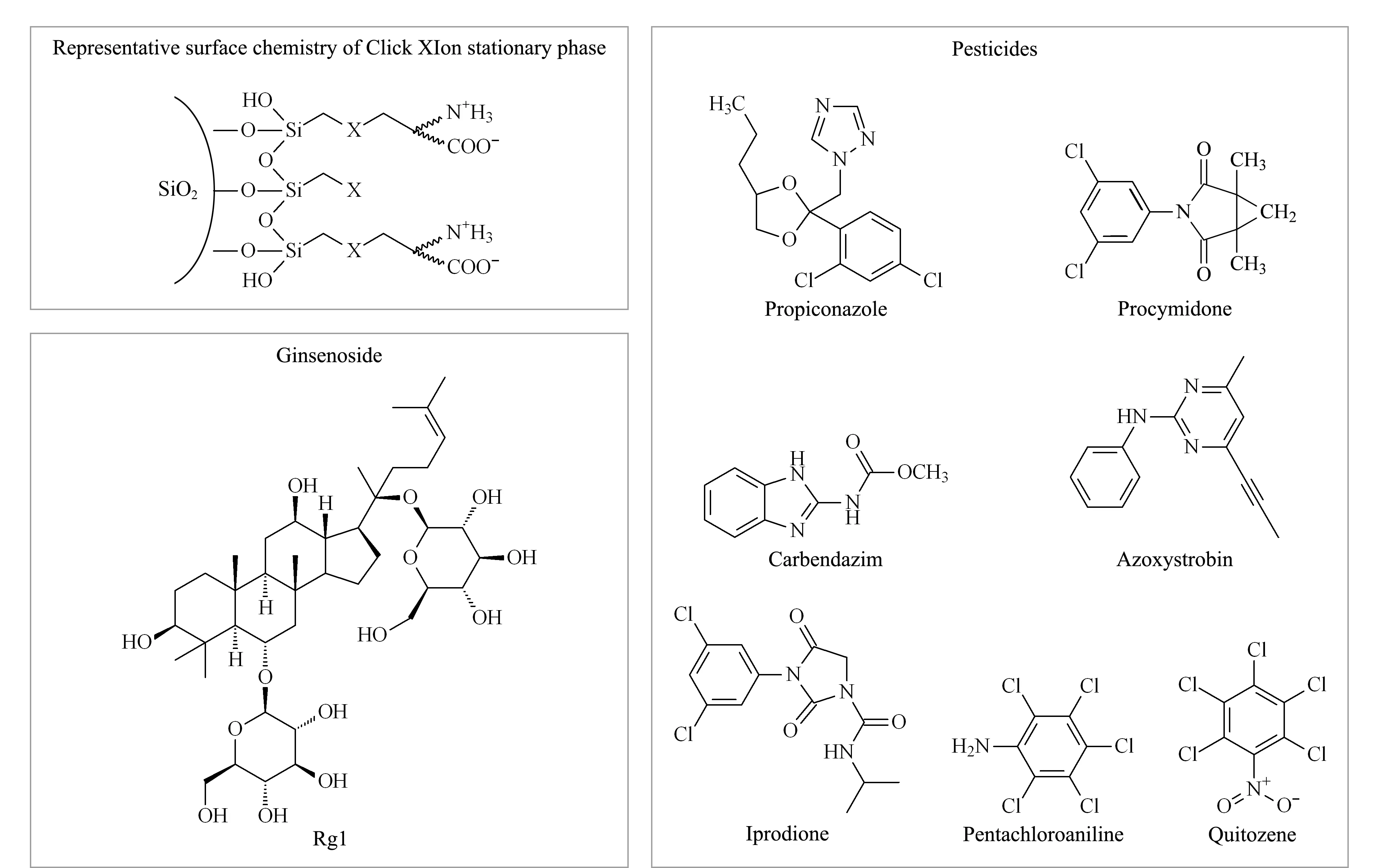
圖 2 Click XIon固定相典型的表面化學(xué)結(jié)構(gòu)和典型人參皂苷及農(nóng)藥結(jié)構(gòu)式Fig.2 Representative surface chemistry of Click XIon stationary phase,structural formulas of typical ginsenoside,and pesticide residues
2.2 農(nóng)殘脫除工藝條件優(yōu)化
使用裝有2 g Click XIon填料(粒徑60 μm)的SPE柱,對(duì)該親水填料脫除農(nóng)殘的最大上樣量、淋洗體積和上樣體積進(jìn)行考察。
2.2.1最大上樣量考察
為減少實(shí)驗(yàn)過(guò)程中皂苷損失,對(duì)最大上樣量進(jìn)行考察。將上述1.4節(jié)中原液Ⅰ上樣,并對(duì)上樣流出液進(jìn)行即時(shí)檢測(cè),每上樣1BV(為便于計(jì)算,統(tǒng)一以填料重量對(duì)應(yīng)的數(shù)值為1BV,如2 g填料的1BV定義為2 mL,300 g填料的1BV定義為300 mL,下同)即對(duì)其相對(duì)應(yīng)的流出液進(jìn)行液相色譜分析,如圖3中b、c、d和e即分別為上樣流出液Ⅰ、Ⅱ、Ⅲ和Ⅳ分析所得色譜圖。圖3a、f、g、h分別對(duì)應(yīng)的溶液為原液Ⅰ、95%乙醇溶液淋洗所得淋洗液、80%乙醇溶液洗脫所得洗脫溶液Ⅰ和60%乙醇溶液洗脫所得洗脫溶液Ⅱ。如圖3e中所示,當(dāng)上樣流出第4BV,即上樣4BV,根據(jù)公式(3)計(jì)算上樣量為10%時(shí),上樣流出液中出現(xiàn)皂苷峰,根據(jù)峰面積計(jì)算,在所上樣皂苷中的含量占比為1.1%,說(shuō)明該上樣量已接近填料載樣的極限,故選擇10%的上樣量做進(jìn)一步的條件優(yōu)化。
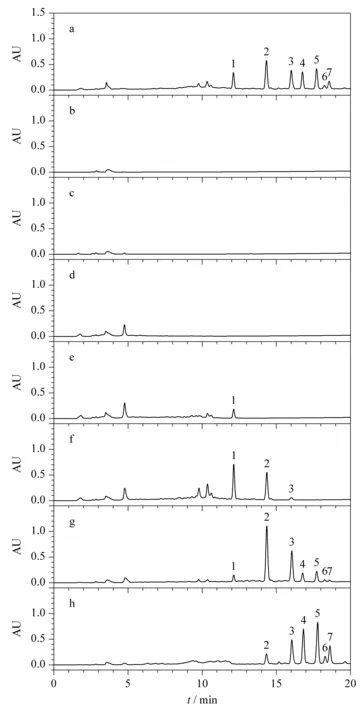
圖 3 最大上樣量考察時(shí)不同餾分的分析色譜圖Fig.3 Chromatograms of different fractions during the investigation of the maximum sample loading mass Mobile phase A:acetonitrile;mobile phase B:water.Elution program for Click XIon (250 mm×4.6 mm,5 μm):0-20 min,90%A-60%A.a.stock solutionⅠ;b.flow-through solution Ⅰ;c.flow-through solution Ⅱ;d.flow-through solution Ⅲ;e.flow-through solution Ⅳ;f.wash solution;g.eluent Ⅰ;h.eluent Ⅱ.Peak identifications:1.Rg1;2.Rf;3.Re;4.Rd;5.Rc;6.Rb2;7.Rb1.
2.2.2淋洗體積考察
根據(jù)上述2.2.1中結(jié)果,將原液Ⅱ以10%總皂苷的上樣量上樣,絕大部分目標(biāo)化合物保留在SPE柱上。如圖4中a為原液Ⅱ分析所得色譜圖,b為原液經(jīng)過(guò)SPE柱后流出的溶液分析所得色譜圖,從圖4可知,在10%上樣量下,農(nóng)藥和人參總皂苷均有部分流穿。為將仍吸附在填料上的農(nóng)藥沖洗下來(lái),同時(shí)保證人參總皂苷不被洗脫,以95%乙醇溶液為淋洗液,以1 mL即0.5BV為單位對(duì)淋洗液體積進(jìn)行依次分析,考察最佳淋洗體積。圖4中c、d、e、f為對(duì)應(yīng)的淋洗液Ⅰ、Ⅱ、Ⅲ和Ⅳ分析所得色譜圖,從圖e中可明顯看出,當(dāng)淋洗體積為1.5 BV時(shí)開始有人參皂苷流出,繼續(xù)淋洗1 mL后,人參總皂苷損失加大,農(nóng)殘峰的峰面積減小。當(dāng)淋洗體積為2 BV時(shí),從圖4g和h中可看出,洗脫餾分色譜圖中無(wú)明顯農(nóng)殘峰存在,此時(shí)根據(jù)公式(1)計(jì)算,農(nóng)殘總脫除率為101.8%,上樣過(guò)程中人參總皂苷損失率為3.5%,淋洗過(guò)程中人參總皂苷損失率為3.9%。根據(jù)農(nóng)殘脫除結(jié)果,選擇淋洗體積為2 BV。
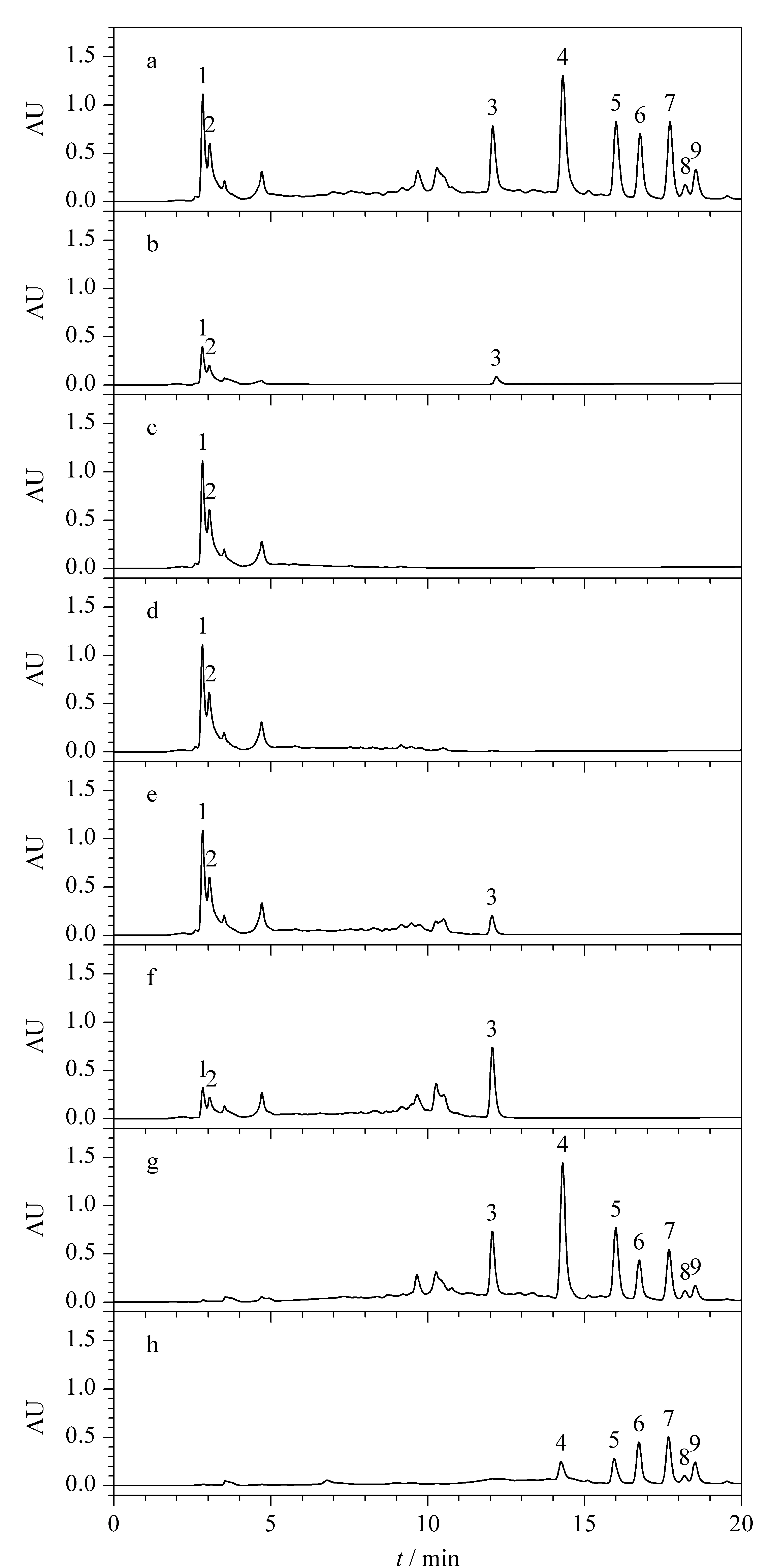
圖 4 淋洗體積考察時(shí)不同餾分的分析色譜圖Fig.4 Chromatograms of different fractions during the investigation of washing solution volumeMobile phase A:acetonitrile;mobile phase B:water.Elution program for Click XIon (250 mm×4.6 mm,5 μm):0~20 min,90%A~60%A.a.stock solution Ⅱ;b.flow-through solution;c.wash solution Ⅰ;d.wash solution Ⅱ;e.wash solution Ⅲ;f.wash solution Ⅳ;g.eluentⅠ;h.eluent Ⅱ.Peak identifications:1,2.pesticide residues;3.Rg1;4.Rf;5.Re;6.Rd;7.Rc;8.Rb2;9.Rb1.
2.2.3上樣體積考察
在上述2.2.1和2.2.2節(jié)中的上樣濃度和體積下,存在部分人參總皂苷穿透現(xiàn)象,可能是由于上樣體積太大造成,故而將上樣液濃度增大,上樣體積減小進(jìn)行SPE小試試驗(yàn)。如圖5中a、b分別為原液Ⅲ和上樣流出液分析所得色譜圖,從圖中可明顯看出,在所確定的最大上樣量和淋洗體積條件下,將上樣體積減小至2 BV,上樣流出液中未出現(xiàn)人參皂苷峰。如圖5c為95%乙醇溶液淋洗所得淋洗液分析所得色譜圖,從圖中可看出人參總皂苷有少量流出損失,經(jīng)計(jì)算,人參總皂苷損失率由7.4%減小至3.4%,且減少了溶劑的使用,提高了效率,在實(shí)際工藝生產(chǎn)中,會(huì)降低生產(chǎn)成本。從洗脫餾分色譜圖5d和e可看出,無(wú)明顯農(nóng)藥峰存在,根據(jù)峰面積計(jì)算農(nóng)藥總脫除率為102.1%,說(shuō)明減少上樣體積在保證農(nóng)殘脫除率情況下,能降低人參總皂苷損失,故最終確定上樣體積為2 BV。
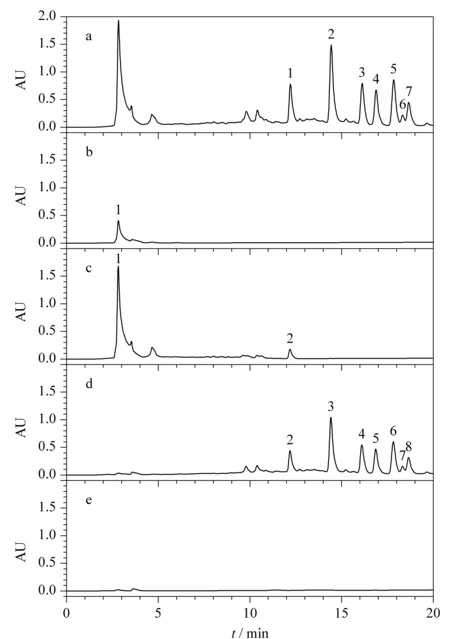
圖 5 上樣體積優(yōu)化時(shí)不同餾分的分析色譜圖Fig.5 Chromatograms of different fractions during the optimization of loading volumeMobile phase A:acetonitrile;mobile phase B:water.Elution program for Click XIon (250 mm×4.6 mm,5 μm):0-20 min,90%A-60%A.a.stock solution Ⅲ;b.flow-through solution;c.wash solution;d.eluentⅠ;e.eluent Ⅱ.Peak identifications:1.pesticide residues;2.Rg1;3.Rf;4.Re;5.Rd;6.Rc;7.Rb2;8.Rb1.
2.3 實(shí)際樣品制備
根據(jù)所優(yōu)化的最佳工藝條件,在DAC50系統(tǒng)上開展放大制備實(shí)驗(yàn),具體條件見上述1.2節(jié),制備譜圖如圖6所示。對(duì)制備所得各餾分進(jìn)行液相色譜分析,根據(jù)公式(1)計(jì)算,具體各餾分中人參總皂苷含量占比如表1所示,合并洗脫液Ⅰ和Ⅱ,根據(jù)公式(2)計(jì)算樣品純度,最終人參總皂苷回收率為94.4%,且純度由59.87%提高到69.61%。

圖 6 人參總皂苷的制備色譜圖Fig.6 Preparative chromatogram of ginsenosides
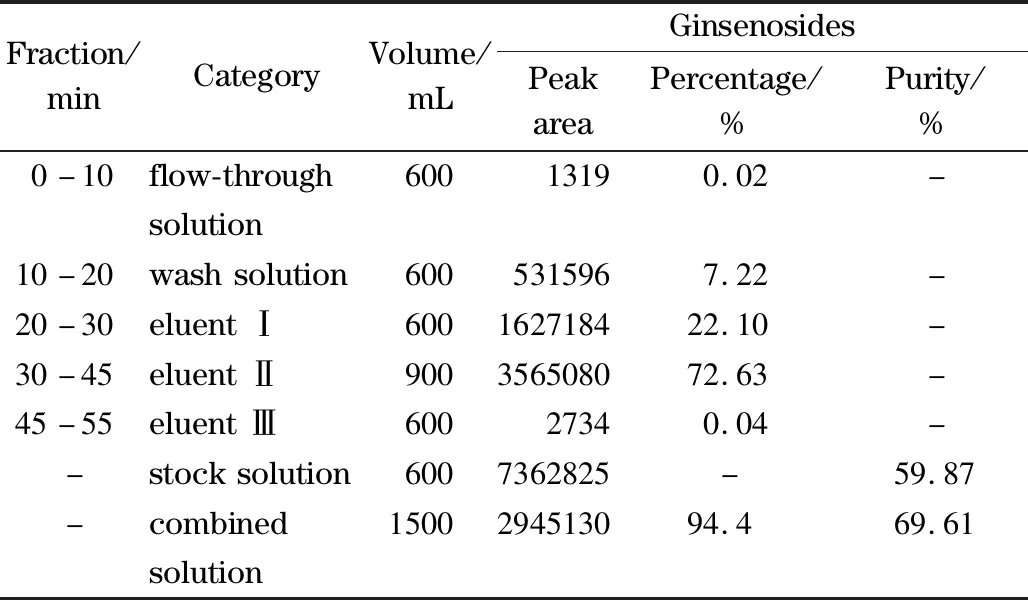
表1 各餾分中人參總皂苷峰面積的百分比及成品純度Table 1 Peak area percentage of total ginsenosides in each fraction and the purity of the finished product
采用GC-MS/MS對(duì)親水色譜脫除前后人參總皂苷樣品進(jìn)行113項(xiàng)農(nóng)殘檢測(cè),參考賽默飛世爾科技對(duì)植物性食品中208種農(nóng)殘檢測(cè)方法包[32],確定113種農(nóng)藥保留時(shí)間及定性和定量離子對(duì),通過(guò)對(duì)上述1.3節(jié)中所配制的混合對(duì)照品溶液進(jìn)行分析,分別繪制出113種農(nóng)藥的標(biāo)準(zhǔn)曲線,所得113種農(nóng)藥的線性相關(guān)系數(shù)(R2)均大于0.99,通過(guò)線性回歸方程對(duì)其進(jìn)行定量計(jì)算。檢測(cè)出脫除前樣品中含有14種含量達(dá)0.1 mg/kg及以上農(nóng)殘,其中皮蠅磷、克百威和甲基毒死蜱含量在0.5 mg/kg以上,經(jīng)親水色譜脫除后,其中9種農(nóng)藥未檢出,另外5種含量降至0.05 mg/kg以下。14種農(nóng)藥脫除前后具體含量見表2,結(jié)果證實(shí)親水色譜對(duì)人參總皂苷中農(nóng)藥殘留具有良好的脫除效果。
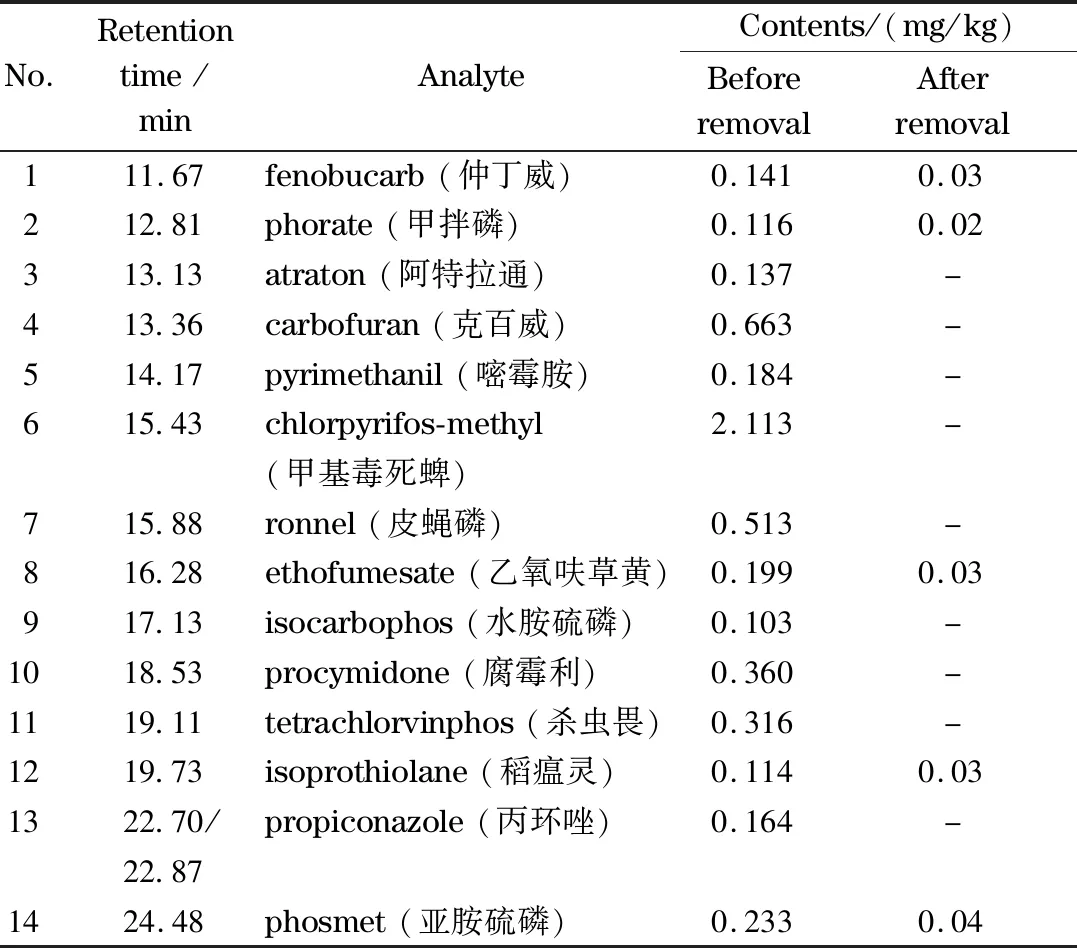
表2 精制樣品中含量在0.1 mg/kg及以上的14種農(nóng)殘脫除前后含量對(duì)比Table 2 Comparison of pesticide contents before and after the removal of 14 pesticide residues in refined samples with contents of 0.1 mg/kg and above
3 結(jié)論
本文以人參提取物為研究對(duì)象,利用人參皂苷與農(nóng)殘之間相對(duì)分子質(zhì)量和親疏水性的差異,考察了人參中常見農(nóng)殘和人參總皂苷在反相和親水色譜柱上的保留行為,確定了親水色譜脫除人參提取物中農(nóng)藥殘留的最佳上樣量和洗脫條件,并放大到制備水平,人參總皂苷回收率達(dá)94%以上且純度提高近10%,對(duì)脫除前后樣品進(jìn)行113項(xiàng)農(nóng)殘檢測(cè),親水色譜脫除前樣品中含量達(dá)0.1 mg/kg及以上農(nóng)殘共14種,經(jīng)親水色譜脫除,所得成品中113種農(nóng)殘均未檢出或含量在0.05 mg/kg以下。與現(xiàn)有的農(nóng)殘脫除方法相比,該方法簡(jiǎn)單、高效、穩(wěn)定,且適用于多種農(nóng)殘脫除,為高品質(zhì)人參及其他中藥提取物產(chǎn)品的開發(fā)及生產(chǎn)提供參考。
