我國東北地區土壤鈾同位素水平、分布和來源
2021-05-29 03:55:32侯小琳張路遠
中國環境科學 2021年5期
黃 釗,侯小琳,趙 雪,張路遠
我國東北地區土壤鈾同位素水平、分布和來源
黃 釗1,3,侯小琳1,2*,趙 雪1,2,張路遠1,2
(1.中國科學院地球環境研究所,黃土與第四紀地質國家重點實驗室,陜西省加速器質譜技術及應用重點實驗室,西安加速器質譜中心,陜西 西安 710061;2.中國科學院第四紀科學與全球變化卓越創新中心,陜西 西安 710061;3.中國科學院大學,北京 100049)
系統采集了我國東北地區132個表層土壤樣品,采用混合酸全溶分解樣品,使用UTEVA萃取色譜分離樣品溶液中的鈾,應用串聯四極桿電感耦合等離子體質譜(ICP-MS/MS)測量樣品溶液中238U、235U、234U,獲得了研究區域表層土壤中3種鈾同位素濃度水平和分布.首次大范圍報道該地區表土中234U水平,發現在部分土壤中234U發生明顯的同位素分餾.235U/238U原子比值分布顯示區域大氣核武器試驗的放射性顆粒物在山脈迎風坡有明顯沉積,導致大興安嶺等山脈的西側235U/238U原子比值較高.但該區域鈾同位素整體上處于環境本底水平,受人類核活動影響較小.來源分析表明研究區域表土中鈾主要來源于成土母巖的巖石風化以及人類的工業和農業活動.
鈾;ICP-MS/MS;環境放射性;土壤;同位素分餾;235U/238U;234U/238U
鈾是天然存在的原子序數最高的元素[1],是核工業的主要原料,隨著鈾礦的開采、核能利用以及核試驗等人類核活動的進行,人為產生的鈾被釋放到土壤等環境介質中[2].鈾同位素具有輻射和化學毒性,研究鈾同位素在環境中的分布、遷移和擴散行為對于環境保護和核安全評估具有十分重要的意義[3-4].鈾元素有15種同位素,質量數從226~240,其中234U(1/2=2.45×105a)、235U(1/2=7.04×108a)、238U (1/2=4.47×109a)是天然放射性同位素,也是最重要的鈾的同位素.天然鈾中豐度最大的為238U,相對豐度為99.27%,235U為0.72%,234U為0.0055%[5].235U作為天然存在的唯一可裂變的鈾同位素,是重要核燃料,235U/238U原子比值可用于評估環境中鈾的水平是否偏離本底水平,是濃縮鈾或者貧鈾污染的判斷依據,以及鈾污染程度的重要指標[6].234U是238U的一個衰變子體,234U/238U的活度比值可用于研究鈾同位素的來源、分布和遷移,以及成礦規律、年齡和鹽湖成因等[7].另外234U還是地質定年的一種重要同位素.分析土壤中的238U、235U、234U濃度以及235U/238U原子比值、234U/238U的活度比值,研究其分布狀況和來源,可用于示蹤環境過程.
目前我國環境樣品中的鈾同位素研究報道較少,天然放射性背景值調查數據僅有少量土壤中天然放射性核素238U、226Ra、232Th和40K等核素的含量[8].這些數據表明土壤中天然放射性核素的分布具有較為明顯的地域性特征,土壤中天然鈾分布與成土母巖有明顯的相關性[9-10].但缺乏系統的、大范圍環境土壤中鈾同位素的研究,特別是攜帶核活動指紋信息(如235U/238U原子比值)的同位素比值數據.土壤中鈾同位素環境過程示蹤研究較少報道.
我國東北地區位于諸多核武器試驗區域和核設施的下風向,特別是前蘇聯塞米巴拉核試驗場、歐洲核燃料后處理廠和我國羅布泊核試驗場等,且礦產資源豐富、重工業產業發達,是我國重要的工業和糧食基地.目前該地區的鈾同位素數據僅有中國環境監測總站1990年發布的中國土壤鈾元素背景值,土壤中鈾同位素238U、235U、234U的濃度以及235U/238U、234U/238U比值尚未見報道,相關研究也較少.因此調查和評價該地區環境放射性水平,研究該地區土壤環境中天然鈾同位素的水平和分布,對于判斷該地區環境是否受到放射性污染、研究放射性核素在環境中的遷移轉化規律以及研究放射性物質在土壤中的環境效應具有重要意義.
在未受污染的土壤樣品中,天然鈾含量較低,小于8mg/kg[11].對于痕量和超痕量的238U、235U、234U測量,相比于傳統的分析方法,如能譜法、中子活化法、原子吸收光譜、激光熒光分析和γ譜儀等[12-13],電感耦合等離子體質譜法不但可以同時測定3種鈾同位素,而且靈敏度高、檢出限低、樣品制備較為簡單并可進行大批量樣品快速測定.
本研究采用混合酸全溶法提取土壤樣品中的鈾,使用UTEVA萃取色譜從樣品溶液中分離和提純鈾,然后應用串聯四級桿電感耦合等離子體質譜儀(ICP-MS/MS)對采集于我國內蒙古和東北地區的132個表層土壤樣品中的238U和235U以及超痕量的234U(<1ng/g)進行測定,獲得該地區表層土壤樣品中天然鈾同位素的濃度及其空間分布特征,研究人類核活動對該區域的影響程度,探討環境中鈾同位素來源及其遷移和擴散行為.
1 材料與方法
于2014~2015年在我國東北三省以及內蒙古、河北、山西、北京等部分地區132個樣點采集土壤樣品(圖1).考慮到人類擾動對采樣點的影響,采樣時盡量選取遠離城鎮地區,且近百年以來未發生過地質或地貌變化以及未經受嚴重的人類活動擾動的地區作為采樣點.對于人類活動高度密集地區,由于農田耕作層一般不超過表層30cm,主要選取農田作為采樣點.使用直徑為5cm的手動鉆孔土壤采樣器,采集土壤柱,收集在塑料袋中,每個采樣點按相距1m的等距離點采集5個平行土壤柱,運回實驗室.樣品在空氣中干燥后破碎,然后再在60?C烘箱中烘干至恒重.將每個采樣點相同深度的5個平行樣品合并和混合.取大約10g土壤于研缽中研磨,過200目篩子備用.
圖1 采樣點和研究區域示意
準確稱取約0.2g表層(0~5cm)土壤樣品和標準土壤樣品GSR-1至耐高溫玻璃燒杯中,于450?C下高溫灼燒12h,除去有機質.將灼燒后的樣品轉移至Teflon坩堝中,加入0.2g已配置好的233U標準溶液(233U的濃度為0.2779ng/g)用做產率示蹤劑,再加入10mL濃HNO3和10mL濃HF.將盛有樣品及消解液的Teflon坩堝置于帶孔電熱板上,加蓋升溫至90?C進行消解約2h,然后升溫至180?C消解至樣品完全溶解.開蓋加熱蒸發至近干,然后用20mL 3mol/LHNO3溶解,用于色譜分離純化.將用水浸泡過后的UTEVA樹脂裝入2mL色譜柱,用10mL 3mol/LHNO3預平衡樹脂柱.將準備好的消解液加載到UTEVA樹脂上,再用20mL 3mol/LHNO3洗滌樹脂去除樣品中的基體元素,然后用30mL 0.01mol/L HNO3洗脫吸附在樹脂上的鈾于50mL的燒杯中.將燒杯中的洗脫液加熱蒸發至近干,最后用3wt%的HNO3轉移并定容于離心管中.
取1000μg/g的238U標準溶液,用3wt%的HNO3溶液逐級稀釋配制成1, 2, 5, 20, 50ng/g的鈾系列標準溶液.使用3wt% HNO3作為儀器清洗液,使用100ng/g的標準鉍溶液做內標溶液.使用三重四極桿ICP-MS/MS(Agilent 8800)測量鈾同位素(234U、235U、238U).使用2ng/g鈾標準溶液調節儀器參數,獲取238U的信號大于8×105cps/(ng/g).測量238U系列標準溶液,制做標準曲線.樣品測量時每8~10個樣品后加入1個空白(3wt% HNO3)和2個鈾標準溶液(2, 5ng/g)校準.每個樣品溶液測量時間約為3min,其中清洗時間約60s,進樣時間90s.
2 結果與討論
2.1 我國東北地區表土中鈾同位素水平及分布
本次研究共分析了132個表土(0~5cm)樣品,獲得了所有樣品中的235U和238U濃度.部分樣品中鈾濃度較低,測得的234U濃度不確定度大于10%,故只準確獲得其中92個樣品中234U濃度(圖2).
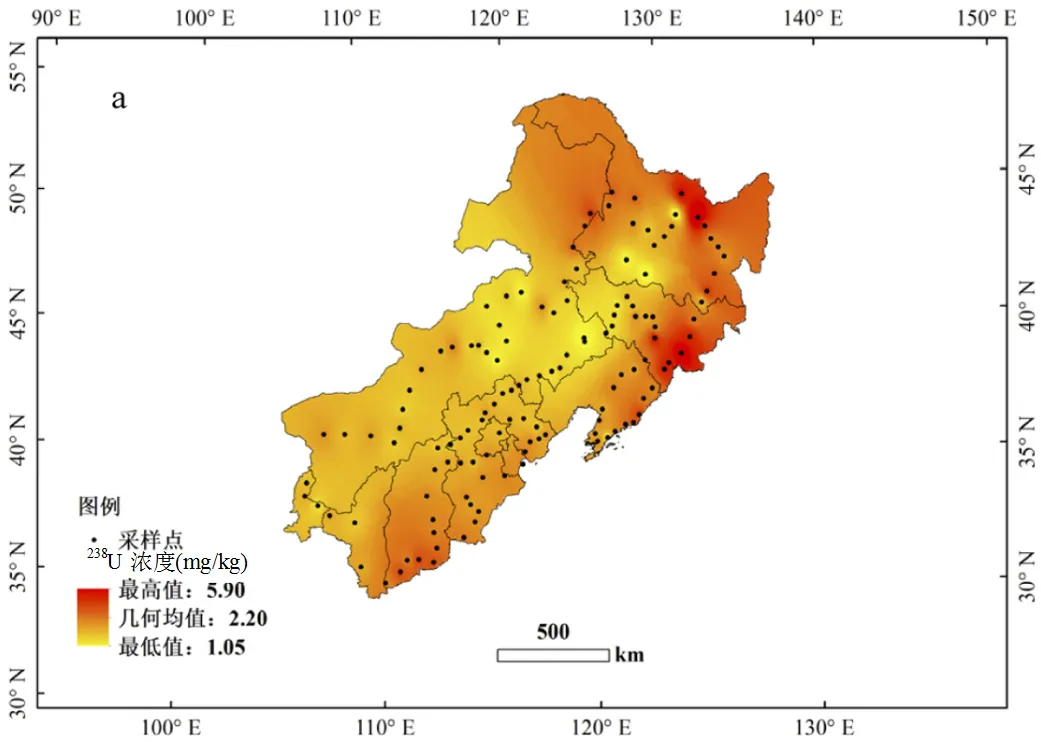
研究區域表層土壤中238U的濃度范圍為1.05~5.90mg/kg,平均值為(2.20±0.70) mg/kg,大部分樣品(>88%)中238U的濃度小于3mg/kg.其中最小238U的濃度((1.05±0.01) mg/kg)位于黑龍江伊春市,最大值((5.90±0.03) mg/kg)位于黑龍江鶴崗市,研究區域的最高值與最低值相差5~6倍.測量得到的總鈾濃度(因238U的天然豐度大于99.3%,故用238U濃度代表總鈾濃度)與中國環境監測總站1990年發表的中國土壤元素背景值[14](0.42~21.1mg/kg,平均值(3.03±1.31) mg/kg)基本吻合,調查區域所得總鈾濃度平均值略低于全國土壤中總鈾濃度平均值(約低23%).研究區域總鈾濃度的分布趨勢為:黑龍江東北部和吉林東部(14個樣品)明顯高于其他區域,濃度范圍為(2.47~5.90) mg/kg,平均值為(3.59±0.94) mg/kg,比該區域的總體平均值高出約57%.另外山西南部、黑龍江與內蒙古交界處略高于其它區域,其余區域為低值區,平均值為(2.14±0.49) mg/kg,比總體平均值低6.5%,且分布較為均勻.低值區分布連續成片,高值區分布則較為零散(圖2a).
研究區域表層土壤中235U的濃度范圍為7.31~ 40.37ng/g,平均值為(15.47±4.82) ng/g,與238U濃度分布相似,最小值為((7.31±0.05) ng/g)位于黑龍江伊春市,最大為((40.37±0.20) ng/g)位于黑龍江鶴崗市,最高值和最低值相差5~6倍,且大部分采樣點(>87%)的235U的濃度低于20ng/g.高值區主要位于黑龍江東北部和吉林東部等地,14個高值樣品的濃度范圍為(17.20~40.37) ng/g,平均值為(24.57±6.51) ng/g,較總體平均值高出約53%,剩余區域為低值區(圖2b).研究區域235U和238U的濃度呈現顯著正相關關系(2>0.99)(圖3),顯示235U和238U兩個同位素有相同的來源,且未發生明顯的同位素分餾現象.

圖3 研究區域表土樣品中 235U和238U濃度的相關分析
研究區域表層土壤中234U的濃度范圍為(0.054~0.393) ng/g,平均值為(0.124±0.057) ng/g.其中234U的濃度最小值(0.054±0.001) ng/g位于黑龍江哈爾濱市,最大值為(0.393±0.005) ng/g位于黑龍江鶴崗市.234U的濃度變化較大,最高值和最低值相差近10倍.在黑龍江東北部和吉林東部等與238U有相同的高值點和濃度變化趨勢,在位于內蒙古巴仁查干呼舒的采樣點也發現較高234U(圖2c),同時研究區域234U和238U的放射性活度的相關性分析呈現2個數據群(圖4),大部分采樣點(>93%)的234U和238U的活度呈顯著的正相關關系,相關系數2=0.957.有6個采樣點中的234U和238U活度與其它點有較大差別,但其中的234U和238U活度也存在顯著的正相關關系,相關系數2=0.942.結果表明,234U和238U在某些區域發生了明顯的同位素分餾現象.
234U是238U的衰變子體,在衰變平衡下,234U/238U放射性比為1.0,在巖石和礦物中,238U衰變過程中產生的反沖作用會使衰變子體234U發生位移,沖出礦物顆粒,會在周圍水溶液的淋濾下進入水體,并通過水體進入周圍土壤,發生鈾同位素分餾現象[7],導致其相對較高的234U/238U比值[1,15].
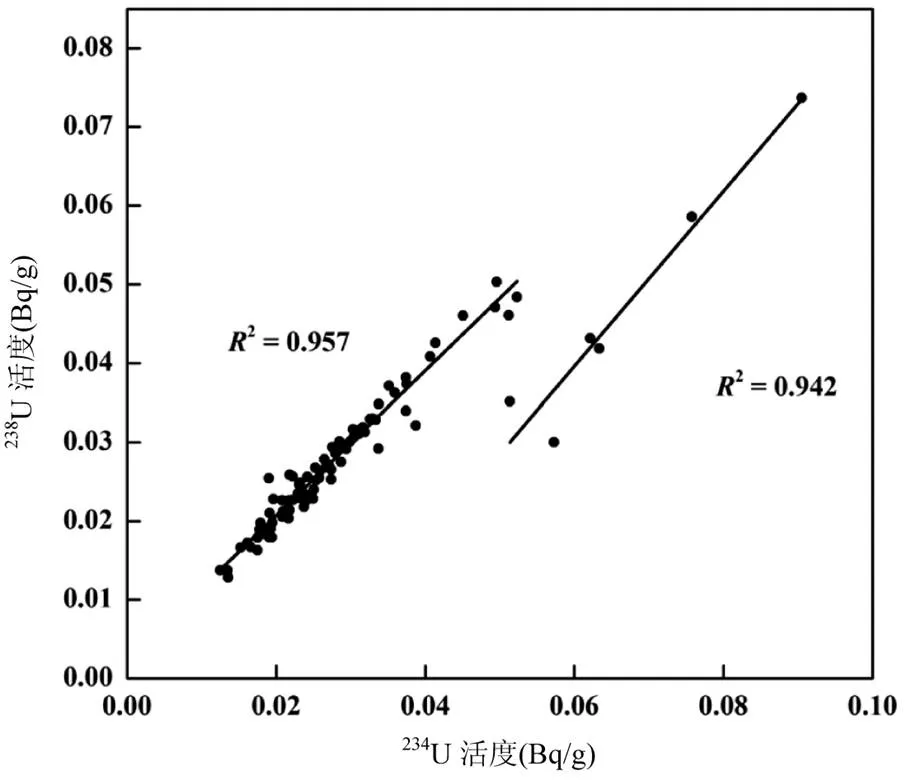
圖4 研究區域表土中234U和238U活度的相關分析
2.2 235U/238U原子比值及分布
2.2.1235U/238U原子比值 研究區域表土中235U/238U原子比值范圍為0.00678~0.00766,平均值為(0.00710±0.00070),天然水平為0.00725[16].最小值為(0.00678±0.00017),高于貧鈾核廢料中235U/238U原子比值(0.004),也高于已報道的受到貧鈾彈污染的巴爾干地區周圍環境樣品中的235U/238U原子比值(0.0046±0.0004)[17].研究區域235U/238U原子比值最高值為(0.00766±0.00002),低于核污染地區和核能工業生產的低濃縮核燃料中235U/238U原子比值(>0.03)[17].以上結果說明研究區域天然鈾同位素未受到明顯的區域污染,而235U/238U原子比值大部分區域(>78%的樣品)略低于天然水平(0.00725),可能是由于核工業活動中開采和235U濃縮活動產生的大量低濃鈾對于地表環境的影響所致[18].
2.2.2235U/238U原子比值的分布及核活動的影響235U/238U原子比值空間分布具有明顯的區域差異,呈現西高東低趨勢,研究區域中的內蒙古中部、山西等區域略高于天然水平(圖5),該區域28個樣品的原子比值范圍為0.00695~0.00766,大部分樣品略高于天然水平0.00725,該結果表明研究區的西部受到人類核活動釋放的含高235U富集鈾的沉降影響.
從235U/238U原子比值的空間分布(圖5)和研究區域圖(圖1)可以發現研究區域內大興安嶺、太行山等山脈的西側235U/238U原子比值較高.研究區域位于北半球西風帶(西風帶范圍為700~200hPa,對應的高度約為3000~10000m),常年盛行西風,并且處于諸多核試驗場和核設施下風向,由于高大山脈的阻隔,人類核活動釋放的富含235U的氣溶膠,可能在高大山脈的迎風坡有較高的沉降,從而對表層土壤中的鈾同位素產生影響,造成在這一區域較高的235U/238U水平.但是由于鈾為非揮發性元素,除了極細顆粒結合態,大部分鈾存在于大顆粒中,傳輸距離有限,主要沉降在核試驗和核設施周圍局部區域.有研究表明,人類核活動會向大氣和氣溶膠中排放鈾,通過大氣沉降影響周圍的環境介質中鈾的同位素組成[6,20-21],所以大部分人類核活動產生的含有高原子比值的鈾同位素在未到達本研究區域之前已經沉降,故研究區域235U/238U原子比值沒有發生明顯的增高,只有少部分細顆粒傳輸到該區域,造成部分地區表土鈾同位素水平略高于天然水平.此外,235U/238U原子比值在該區域西高東低的空間分布與報道的人工129I空間分布一致[22-23],證明了人類核活動沉降對我國東北和內蒙古地區表土鈾同位素的影響.
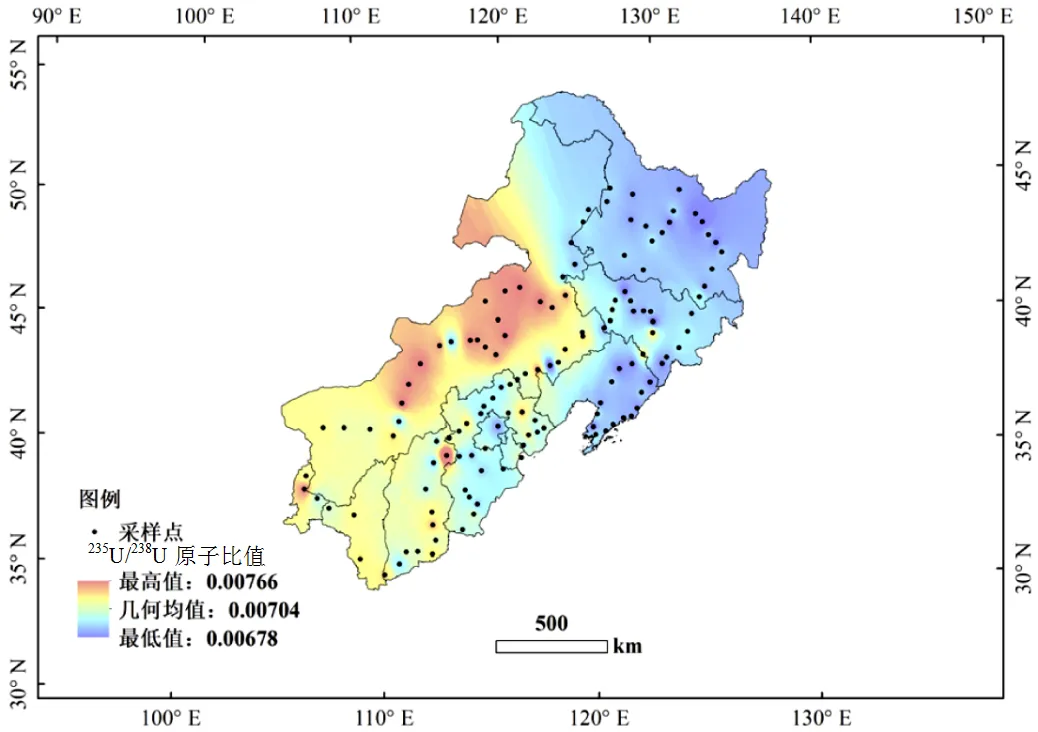
圖5 研究地區表土中235U /238U原子比值空間分布
核設施在正常運行時向環境釋放的極少量含鈾氣溶膠傳輸的高度和范圍有限,以局部沉降為主,不會引起大范圍235U/238U原子比值整體發生改變,故影響該地區表土中鈾同位素分布主要是核事故釋放到大氣中的放射性物質沉降和20世紀80年代以前的大氣核武器試驗的大氣沉降.迄今對全球影響最大、可能對研究區域產生影響的核事故有發生于1986年切爾諾貝利(距離研究區域約6000km)和2011年福島(距離研究區域約1000km)的核電廠事故.與钚等錒系金屬元素的化學性質相似,鈾也是親顆粒性重金屬元素,在大氣中主要以氣溶膠的形式擴散.大量研究表明切爾諾貝利核事故釋放的放射性物質主要沉降在歐洲區域,在我國境內切爾諾貝利事故的放射性沉降極小[24],目前尚未在東亞地區發現切爾諾貝利核事故钚信號,因此該事故對研究區域表層土壤中的鈾同位素貢獻可以忽略不計.福島核事故發生地點雖然離我國較近,但是研究表明,僅在事故點周圍30km內的部分樣品中發現福島钚信號[25],因此福島核事故對距離超過1000km的研究區域的表層土壤中的鈾同位素的影響也可忽略不計.
全球核武器試驗,特別是大當量試驗產生的大部分放射性物質進入平流層,經過長時間混合后沉降到地表,造成全球沉降.但這類沉降較小,不會因地形地貌而導致235U/238U原子比值的差異,故全球大氣沉降對于研究區域235U/238U原子比值分布影響較小.除進入平流層的放射性物質,還有一部分放射性物質僅進入對流層,導致大氣核試驗放射性物質的區域沉降.靠近研究區域的核試驗場有前蘇聯塞米帕拉金斯克核試驗場和我國羅布泊核試驗場,分別進行了86次和23次的大氣核武器試驗,以及30次和5次地面核試驗,其中進入對流層的放射性物質高達0.67Mt[26].由于核試驗場以及我國北部盛行西風,進入對流層的放射性物質主要通過西風傳輸和擴散,西風帶底界高度是3000m.已有研究表明,塞米帕拉金斯克大氣核武器試驗的區域沉降,被西風載帶,大部分放射性物質向東擴散并沉降[27].我國新疆北部土壤中钚同位素分析表明塞米帕拉金斯克核試驗場釋放的钚是其一個重要來源[28].通過HYSPLIT模型對1954年10月30日地面核試驗后的氣團前向運行軌跡的模擬顯示核試驗后3000m左右高空的放射性煙云可將攜帶鈾的細顆粒傳輸至研究區域(圖6),該煙云在到達大興安嶺西麓的時候下降至500m左右,低于大興安嶺的平均海拔1000m.同理,我國羅布泊核試驗場的大氣核武器試驗產生的放射性煙云在中間沒有高大山脈阻隔的情況下也能通過河西走廊將攜帶鈾的細顆粒傳輸至研究區域.有研究表明,重金屬等污染物在山地或者高原的迎風坡富集或者在山區冷阱的作用下沉降[29-30].核試驗產生的攜帶鈾的細顆粒在大興安嶺等高大山脈的阻隔下,通過沉降進入土壤,從而對研究區域表土中鈾同位素產生影響,造成在這一區域較高的235U/238U比值.除大氣核試驗排放的直接擴散和沉降外,沉降在我國西北部和北部的細顆粒,由于氣候影響可能再懸浮,并通過西風作用向研究區域傳輸再沉降.

圖6 1954年10月30日塞米帕拉金斯克核試驗后的氣團運行軌跡[31]
2.3 表土中234U的分布和來源
研究區域234U/238U原子比值的范圍為4.55× 10-5~10.25×10-5(活度比值范圍為0.75~1.91),平均值(5.52±0.78)×10-5(活度比值平均值為1.02±0.14),與天然水平5.54×10-5(活度比1.00)一致,但變化范圍較大.在個別地區出現較高的234U/238U活度比值,但沒有明顯的地理分布特征,高值點主要分布在內蒙古東北部、黑龍江和吉林東部(圖7).57%的樣品點表土中的234U/238U活度比在0.8~1.2, 34%的樣品點活度比小于0.8.前文提到的234U和238U活度存在另一種顯著的正相關關系, 6個樣品點的234U/238U活度比也較高,高于天然水平的1.0(表1).
造成研究區域土壤環境中234U/238U原子比值和活度比值變化較大的原因主要是由于自然過程導致的同位素分餾,使得234U在土壤環境中流失或者富集.導致234U在土壤環境中流失的因素可能為地形地勢的影響.有研究發現平原地區土壤和巖石中234U豐度較低,234U/238U活度比低于1.0[32].這是由于238U在衰變過程中由于反沖作用將234U彈射出土壤小顆粒,隨土壤溶液和地下水侵蝕以及長期的風化,234U從土壤中流失[33].本研究發現平原地區大部分土壤中234U/238U活度比小于0.8(圖7).
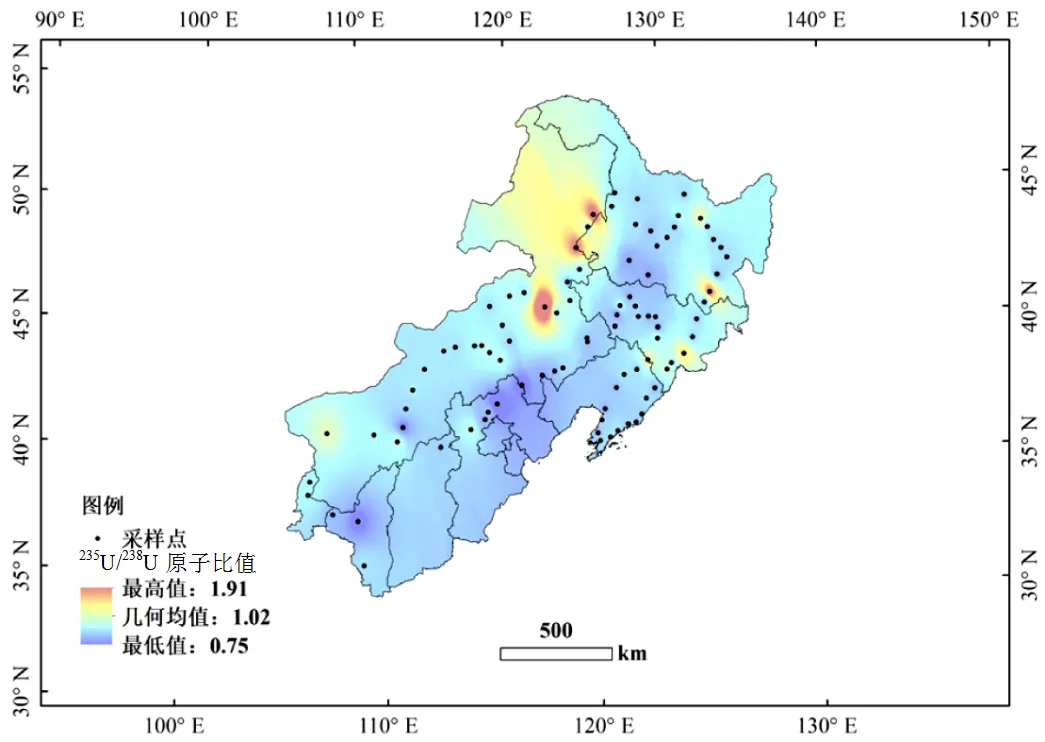
圖7 研究地區表土中234U /238U活度比值空間分布
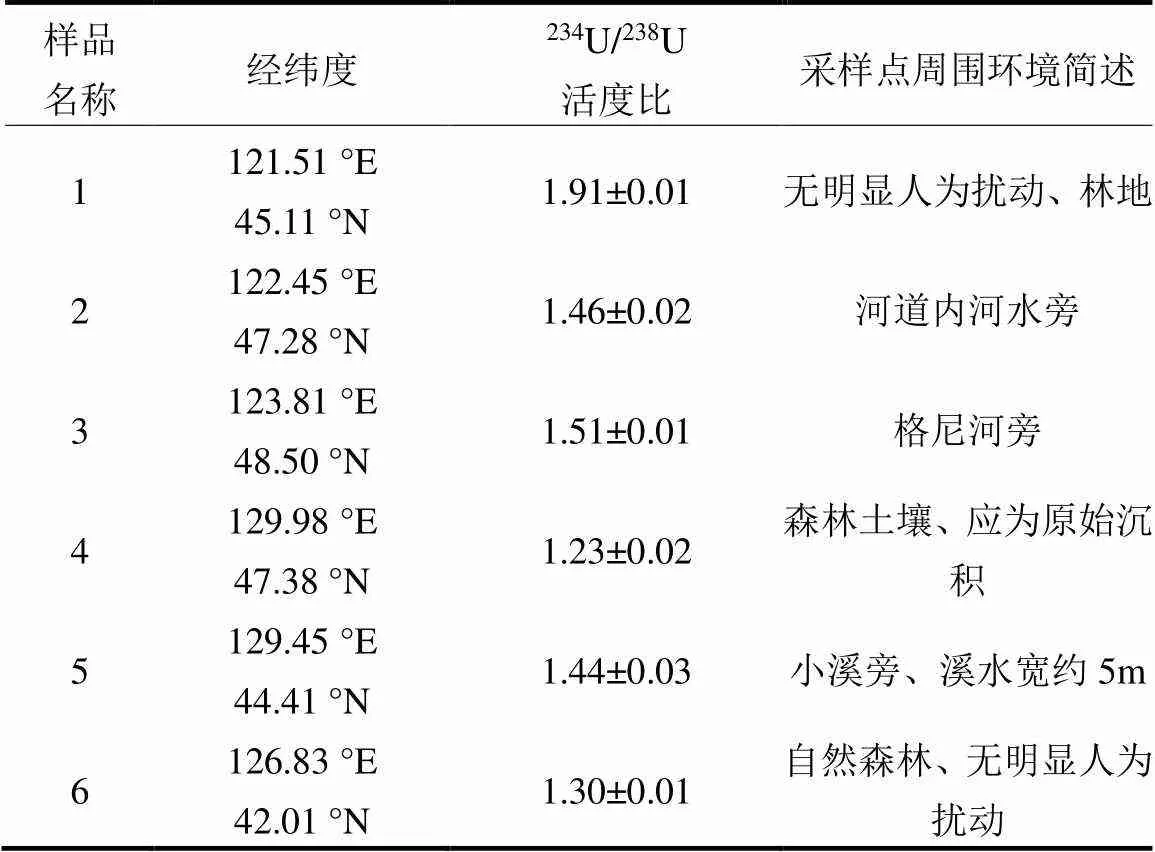
表1 234U/238U活度比大于1.2的樣品信息
有研究表明在各類水體中234U明顯富集,234U/238U活度比大于1.0.不同巖石類型的地下水中234U/238U活度比差別較大.花崗巖地區地下水有比較高的234U/238U活度比值,在山區,由于地表水不斷沖刷,將巖石和礦物表面的鈾沖刷進水體,故山區地表水中234U/238U活度比較高[32].本研究中234U明顯富集的樣品(表1)采樣點的成土母巖均為花崗巖.3個采樣點位于河流附近,其余3個則位于含水量較高的林地,其中1~3號樣品采集于大興安嶺山區,4~6號樣品采集于東北平原,地表水和地下水富含234U,滲入土壤,導致土壤中234U明顯富集.各類水體中234U主要來自于周圍環境介質中,如巖石礦物中234U的有效浸出,花崗巖中鈾含量一般較高,因此遷移至表層土壤中的234U量可能較多.被包裹在巖石和礦物質中的238U通過衰變至234Th,衰變時釋放的能量主要轉移到重核衰變子體234Th上,通過反沖作用使得234Th可能移出巖石或礦物晶體,進入裂隙或者礦物顆粒表面,短壽命的234Th(1/2=24.1d)衰變至另一短壽命的234Pa(1/2=6.7h),并進一步衰變成長壽命的234U(1/2=2.45×105a)[7].進入巖石和礦物質裂隙或顆粒表面的234U與孔隙水和地下水作用,可與其中的碳酸鹽形成水溶性絡合物,通過各類地表水將234U載帶至表層環境,并擴散進入表層土壤中,從而改變土壤中234U的豐度.
2.4 土壤環境中鈾的來源
土壤環境中鈾同位素的來源主要有成土母巖、地表水轉運、人為工農業活動如磷礦的開采、煤礦的開采、磷肥的使用以及人類核活動排放到大氣中的同位素沉降等[9,34].另外土壤中鈾同位素的分布和遷移還和鈾的形態以及土壤的理化性質有關.
研究區域中235U/238U原子比值數據表明該地區鈾同位素主要為天然來源,雖然位于上風向的大氣核試驗一定程度地影響了內蒙古和山西中部地區的235U/238U原子比值,但是對總鈾濃度影響極小,故人類核活動不是影響該地區表土中鈾水平和分布的主要因素.天然水體中的鈾元素濃度基本小于4μg/L[35-37],遠低于表層土壤中鈾元素的含量(3mg/ kg).因此降水和地表水對于表層土壤中總鈾的貢獻有限.故該地區表土中總鈾的濃度主要與成土母巖和人類工業和農業活動有關.
2.4.1 成土母巖的影響 鈾元素經歷地質過程在巖石中積累,其中花崗巖富含鈾元素,其它巖石中如砂巖、沖積巖等鈾含量明顯低于花崗巖,我國花崗巖中鈾濃度為4~30mg/kg[38].在土壤形成過程中,成土母巖風化再沉積,如花崗巖中的鈾同位素伴隨著巖石風化的過程進入土壤,成為土壤礦物質的組成元素.研究區域表土中238U濃度分布(圖2a)和該地區各類巖石中鈾分布基本吻合,富含鈾的花崗巖主要分布于黑龍江東北地區、吉林東部和大興安嶺地區,其它地區的巖石則以砂巖和沖積巖等為主[39].這表明研究區域的鈾可能主要來源為成土母巖巖石風化過程.
2.4.2 采礦和燃煤等工業活動的影響 有研究表明我國各類煤碳中鈾的含量均較高,約為3~20mg/kg[40].研究區域中有黑龍江鶴崗、雞西、吉林遼源、遼寧阜新、撫順等眾多大型煤田,該研究區域表土高鈾區域與這些煤田的分布基本吻合.煤礦開采和燃煤等工業活動產生的飛灰和細顆粒會通過大氣擴散至周圍區域,導致環境中天然放射性水平增加[41-42].含鈾大氣顆粒物通過沉降進入土壤,增加研究區域表層土壤中鈾水平.故采礦和燃煤等工業活動可能是該區域表土中鈾同位素的一個重要來源.
2.4.3 土壤中鈾的保留 分析結果表明,研究區域表土中鈾濃度與土壤有機質含量呈顯著性正相關(=0.656,<0.01)(圖8).其它研究也發現了土壤有機質含量和鈾含量的正相關性[43-44].有機質的含量影響土壤氧化還原環境,從而影響土壤中鈾的存在形態及遷移[45].高有機質含量的土壤環境易呈還原性環境,高流動性U(VI)被還原成親顆粒的U(IV),有利于鈾在土壤中的滯留.反之,有機質含量較低時,鈾主要以鈾酰離子的形式存在,易于與土壤溶液中的碳酸根形成水溶性絡合物,從原有土壤中淋濾,進入水體而遷移[43].因此土壤有機質是土壤中鈾保存的關鍵因素,影響表土鈾水平.
2.4.4 其它影響土壤鈾的因素 現代農業中大量使用化肥,其中磷肥和土壤改良劑的使用可能是該區域表土中鈾同位素的另一個來源.由于磷、鈾共生的特點,使得鈾經常與磷礦伴生.研究表明磷肥中鈾元素的含量高達1.3mg/kg[10].東北地區是我國主要農業生產區,大部分地區均已開墾,很難找到完全未受人為干擾的采樣點,大部分采集點位于農田附近.伴隨磷肥的大量施用,其中的鈾進入土壤中,并在土壤環境中進行遷移轉化.
另外,20世紀60年代東北的開荒過程中,粉煤灰作為土壤改良劑大范圍使用.有報道粉煤灰施用量的增加也會導致土壤中天然放射性活度明顯升高[46].故磷肥的施用和粉煤灰等土壤改良劑的使用可能是影響該區域表土中鈾同位素的分布和水平的一個重要因素.但由于其使用量有限,磷肥和粉煤灰中鈾濃度與土壤中鈾濃度相當或稍高,因此其可能不是土壤中鈾的主要來源.
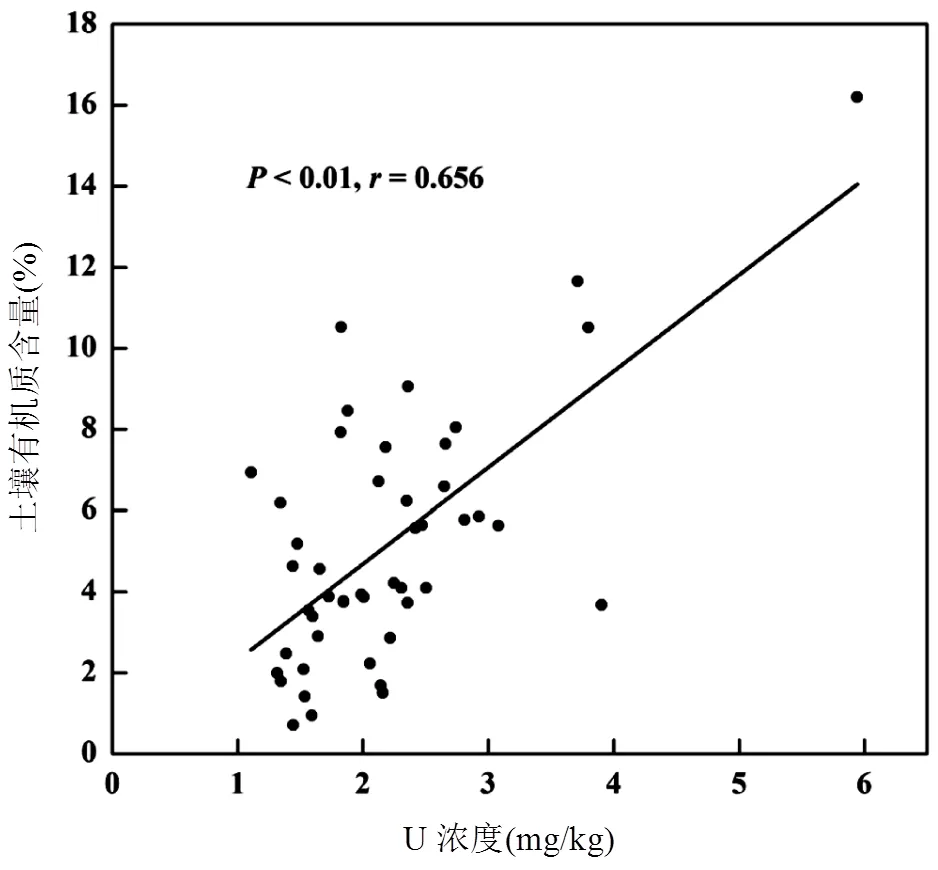
圖8 表層土壤樣品中鈾濃度和土壤有機質的關系
3 結論
3.1 我國東北地區表土中鈾含量基本處于天然環境本底水平,影響其水平和分布的因素主要有成土母巖的巖石風化和再沉積、采礦和燃煤等工業活動、土壤有機質含量以及農業所用的磷肥和土壤改良劑的施用等,其中成土母巖的巖石風化過程是影響其水平和分布的最主要因素.高值區主要位于黑龍江東北部和吉林東部等花崗巖分布較多、煤田煤礦分布較多的區域.
3.2 研究區域表層土壤中235U和238U的分布具有極好的相關性,未發生明顯的同位素分餾現象.234U濃度以及234U/238U活度比值范圍較大,在一些采樣點出現較高的234U/238U活度比值.這主要是由于238U衰變中產生的子體在反沖作用下從巖石和礦物顆粒內部進入表面和裂隙,通過淋濾進入水體,導致較高的234U/238U活度比值,出現明顯的同位素分餾現象.
3.3 研究區域表層土壤中235U/238U原子比值基本處于天然水平(0.00725),受人類核活動影響較小.研究區域表土中235U/238U原子比值呈現西高東低的趨勢,這主要歸因于核活動污染的氣載顆粒物沉降受地形影響.位于塞米帕拉金斯克和我國羅布泊的核試驗場在20世紀40~80年代進行的大氣核武器試驗釋放了大量235U,含鈾細顆粒在西風環流的作用下向東擴散,在大興安嶺高山的阻隔下在西麓出現較高的沉降,導致該區域235U/238U原子比值明顯高于東北平原.
[1] Skwarzec B, Bory?o A, Struminska D.234U and238U isotopes in water and sediments of the southern Baltic [J]. Journal of Environmental Radioactivity, 2002,61:354–363.
[2] 景稱心,孔秋梅,馮志剛.中國南方某鈾尾礦庫周緣土壤重金屬污染研究 [J]. 中國環境科學, 2020,40(1):338–349. Jing C X, Kong Q M, Feng Z G. Heavy metal pollution in a uranium mining and metallurgy area in South China [J]. China Environmental Science, 2020,40(1):338–349.
[3] Bai J, Yao H, Fan F L, et al. Biosorption of uranium by chemically modified Rhodotorula glutinis [J]. Journal of Environmental Radioactivity, 2010,101(11):969–973.
[4] Wang J J, He B H, Wei X Y, et al. Sorption of Uranyl Ions on TiO2: Effects of pH, contact time, ionic strength, temperature and HA [J]. Journal of Environmental Sciences, 2019,75:115–123.
[5] Ellis-Akovali Y. Nuclear data sheets for A=234 [J]. Nuclear Data Sheets, 1983,40(4):523–603.
[6] Pourcelot L, Masson O, Renaud P, et al. Environmental consequences of uranium atmospheric releases from fuel cycle facility: II. The atmospheric deposition of uranium and thorium on plants [J]. Journal of Environmental Radioactivity, 2015,141:1–7.
[7] Fleischer R L, Raabe O G. Recoiling alpha-emitting nuclei. Mechanisms for uranium-series disequilibrium [J]. Geochimica Et Cosmochimica Acta, 1978,42(7):973–978.
[8] 曹龍生,楊亞新,張 頁,等.中國大陸主要省份土壤中天然放射性核素含量分布規律研究[J]. 東華理工大學學報(自然科學版), 2012, 35(2):167–172. Cao L S, Yang Y X, Zhang Y, et al. Distribution pattern of radionuclides in the soil of mainland China [J]. Journal of East China Institute of Technology(Natural Science), 2012,35(2):167–172.
[9] Borylo A. Determination of uranium isotopes in environmental samples [J]. Journal of Radioanalytical and Nuclear Chemistry, 2013, 295(1):621–631.
[10] Merkel B, Hasche-Berger A. Uranium in the environment: Mining impact and consequences [M]. Germany: Springer-Verlag Berlin Heidelberg, 2006:57–69.
[11] Meinrath A, Schneider P, Meinrath G. Uranium ores and depleted uranium in the environment, with a reference to uranium in the biosphere from the Erzgebirge/Sachsen, Germany [J]. Journal of Environmental Radioactivity, 2003,64(2/3):175–193.
[12] 侯小琳,王 珂.微量鈾的快速超熱中子活化法測定[J]. 核技術, 1997,20(9):568–571. Hou X L, Wang K. Determination of trace uranium with fast epithermal neutron activation analysis [J]. Nuclear Techniques, 1997, 20(9):568–571.
[13] El-Taher A. INAA and DNAA for uranium determination in geological samples from Egypt [J]. Applied Radiation & Isotopes, 2010,68(6):1189–1192.
[14] 中國環境監測總站.中國土壤元素背景值 [M]. 北京:中國環境科學出版社, 1990:18. China National Environmental Monitoring Centre. Background value of soil element in China [M]. Peking: China Environmental Press, 1990:18.
[15] Skwarzec B, Bory?o A, Struminska D. Activity disequilibrium between234U and238U Isotopes in Southern Baltic [J]. Water, Air, and Soil Pollution, 2004,159(1):165–173.
[16] Alamelu D, Aggarwal S K. Determination of235U/238U Atom ratio in uranium samples using liquid scintillation counting (LSC) [J]. Talanta, 2009,77(3):991–994.
[17] Carvalho F P, Oliveira J M. Uranium isotopes in the Balkan’s environment and foods following the use of depleted uranium in the war [J]. Environment International, 2010,36(4):352–360.
[18] Brennecka G A, Borg L E, Hutcheon I D, et al. Natural variations in uranium isotope ratios of uranium ore concentrates: Understanding the238U/235U fractionation mechanism [J]. Earth & Planetary Science Letters, 2010,291(1–4):228–233.
[19] Warneke T, Croudace I W, Warwick P E, et al. A new ground-level fallout record of uranium and plutonium isotopes for northern temperate latitudes [J]. Earth & Planetary Science Letters, 2002, 203(3/4):1047–1057.
[20] Pettersson H B L, Holm E. Investigation of aerial dispersion of uranium isotopes from a nuclear fuel fabrication facility [J]. Waste Management, 1992,12(1):85–97.
[21] Pourcelot L, Boulet B, Le Corre C, et al. Isotopic evidence of natural uranium and spent fuel uranium releases into the environment [J]. Journal of Environmental Monitoring Jem, 2011,13(2):355–361.
[22] 范煜坤.我國表土中129I的空間分布及129I年代方法學初探[D]. 北京:中國科學院大學, 2013:40–48. Fan Y K. Spatial distribution of129I in Chinese surface soil and preliminary study on the129I chronology [D]. Peking: University of Chinese Academy of Sciences, 2013:40–48.
[23] 魯 彤.中國內蒙古-東北地區陸生植物中129I和鈾同位素分析及環境示蹤[D]. 北京:中國科學院大學, 2018:29–52. Lu T. Investigation and Environmental tracing of129I and uranium isotopes in terrestrial vegetation samples from Inner Mongolia and Northeast China [D]. Peking: University of Chinese Academy of Sciences, 2018:29–52.
[24] Hirose K, Igarashi Y, Aoyama M, et al. Recent trends of plutonium fallout observed in Japan: plutonium as a proxy for desertification [J]. Journal of Environmental Monitoring Jem, 2003,5(2):302–307.
[25] Zheng J, Togami K, Watanabe Y, et al. Isotopic evidence of plutonium release into the environment from the Fukushima DNPP accident [J]. Scientific Reports, 2012,2(3):3041–3048.
[26] Source and effects of ionizing radiation (2000). ANNEX C: Exposures to the public from man-made sources of radiation [R]. Vienna: United Nations Scientific Committee on the Effects of Atomic Radiation, 2000:158-176.
[27] Sukhorukov F V, Gavshin V M, Malikova I N, et al. Cesium-137 in the environment of the Altay Region (Russia) [J]. Water Air & Soil Pollution, 2000,118(3/4):395–406.
[28] Zhao X, Qiao J, Hou X L. Plutonium isotopes in Northern Xinjiang, China: Level, distribution, sources and their contributions [J]. Environmental Pollution, 2020,265:114929.
[29] Dong Z, Kang S C, Qin X, et al. New insights into trace elements deposition in the snow packs at remote alpine glaciers in the northern tibetan plateau, China [J]. Science of the Total Environment, 2015,529:101–113.
[30] Westgate J, Wania F. Model-based exploration of the drivers of mountain cold-trapping in soil [J]. Environmental Science Processes & Impacts, 2013,15(12):2220–2232.
[31] Air Resources Laboratory. HYSPLIT model [EB/OL]. https://www. ready.noaa.gov/HYSPLIT.php.
[32] 劉鐵庚.天然鈾同位素的某些地球化學特征 [J]. 地質地球化學, 1979,12:44-51. Liu T G. Some geochemical characteristics of natural uranium isotopes [J]. Earth & Environment, 1979,12:44–51.
[33] Lee V, DePaolo D, Christensen J. Uranium-series comminution ages of continental sediments: Case study of a Pleistocene alluvial fan [J]. Earth and Planetary Science Letters, 2010,296:244–254.
[34] Borylo A, Skwarzec B. Bioaccumulation of polonium (210Po) and uranium (234U,238U) in plants around phosphogypsum waste heap in Wi?linka (northern Poland) [J]. Radiochimica Acta, 2011,99(11):719–731.
[35] Mangini A, Sonntag C, Bertsch G, et al. Evidence for a higher natural uranium content in world rivers [J]. Nature, 1979,278:337–339.
[36] Nriagu J, Nam D, Ayanwola T A, et al. High levels of uranium in groundwater of Ulaanbaatar, Mongolia [J]. Science of the Total Environment, 2012,414:722–726.
[37] Favas P, Pratas J, Mitra S, et al. Biogeochemistry of uranium in the soil-plant and water-plant systems in an old uranium mine [J]. Science of the Total Environment, 2016,568(10):350–68.
[38] 牟保磊.元素地球化學[M]. 北京:北京大學出版社, 1999:124-125. Mou B L. Elemental geochemistry [M]. Peking: Peking University Press, 1999:124-125.
[39] 中國地質調查局.地質圖[EB/OL]. http://www.cgs.gov.cn. China Geological Survey. Geological map [EB/OL]. http://www.cgs. gov.cn.
[40] 黃文輝,唐修義.中國煤中的鈾、釷和放射性核素[J]. 中國煤炭地質, 2002,14:55–63. Huang W H, Tang X Y. Uranium, thorium and radionuclides in Chinese coal [J]. Coal Geology of China: 2002,14:55–63.
[41] Tsikritzis L, Ganatsios S, Duliu O, et al. Natural and artificial radionuclides distribution in some lichens, mosses, and trees in the vicinity of lignite power plants from west Macedonia, Greece [J]. Journal of Trace and Microprobe Techniques, 2003,21(3):543–554.
[42] Haribala, Hu B, Wang C, et al. Assessment of radioactive materials and heavy metals in the surface soil around uranium mining area of Tongliao, China [J]. Ecotoxicology & Environmental Safety, 2016, 130(8):185–192.
[43] 張鐘先,田均良.中國黃土地區土壤中天然放射性元素的生物地球化學[J]. 土壤學報, 1995,32(4):353–361. Zhang Z X, Tian J L. Biogeochemistry of natural radioactive elements in soil in loess region of China [J]. Acta Pedologica Sinica, 1995, 32(4):353–361.
[44] 況潤元,汪永進,張向華,等.石筍鈾同位素組成對土壤環境變化的指示[J]. 科學通報, 2002,47(13):1022–1026. Kuang R Y, Wang Y J, Zhang X H, et al. The indication of uranium isotopic composition of stalagmites to soil environmental change [J].Chinese Science Bulletin, 2002,47(13):1022–1026.
[45] 裴晶晶,胡 南,張 輝,等.鈾尾礦中不同形態鈾釋放的影響因素及其相關性[J]. 中國環境科學, 2019,37(9):3073-3080. Pei J J, Hu N, Zhang H, et al.An analysis of influencing factors on the release of different species of uranium from uranium tailings and their correlation [J]. China Environmental Science, 2019,37(9):3073-3080.
[46] 楊俊誠,朱永懿,陳景堅,等.粉煤灰的農業利用及其環境放射性污染評價[J]. 核農學報, 1999,13(5):299–304. Yang J C, Zhu Y Y, Chen J J, et al. Agricultural utilization of coal ash and evaluation of environmental radioactive pollution [J].Journal of Nuclear Agricultural Sciences, 1999,13(5):299–304.
Uranium isotopes in surface soil in Inner Mongolia and northwest China: level, distribution and sources.
HUANG Zhao1,3, HOU Xiao-lin1,2*, ZHAO Xue1,2, ZHANG Lu-yuan1,2
(1.Institute of Earth Environment, Chinese Academy of Sciences, State Key Laboratory of Loess and Quaternary Geology, Shaanxi Key Laboratory of Accelerator Mass Spectrometry Technology and Application, Xi’an AMS Center, Xi’an 710061, China;2.CAS Center of Excellence in Quaternary Science and Global Change, Xi’an 710061, China;3.University of Chinese Academy of Sciences, Beijing 100049, China)., 2021,41(5):2343~2351
In total 132 surface soil samples were collected from Northeast China. The ground and homogenized soil was completely dissolved using mixed acids. Uranium was separated from the sample matrix by extraction chromatography using UTEVA resin, and uranium isotopes in the separated sample were then measured by a triple quadrupole Inductively Coupled Plasma Mass Spectrometry (ICP-MS/MS) to learn the level and distribution of238U,235U,234U concentrations in surface soil of the study. The234U in surface soil of this area was reported first time, and an obvious isotope fractionation of234U was observed in some soil samples indicated by the increased234U/238U ratios.The distribution of235U/238U atomic ratios in these surface soil samples showed an obvious regional deposition of uranium derived from atmospheric nuclear weapon tests the windward slope of the mountain. Consequently, the level of235U/238U atomic ratios in the surface soil in west site of Greater Khingan Mountain were higher than other places. The level of uranium isotopes in surface soil was comparable to the environmental background level in other places, indicating negligible impacts from human nuclear activities. The source analysis showed that uranium isotopes in surface soil were dominated by rock weathering, human industrial and agricultural activities also contributed moderately in some areas.
Uranium;ICP-MS/MS;environmental radioactivity;soil;isotope disequilibrium;235U/238U;234U/238U
X53
A
1000-6923(2021)05-2343-09
黃 釗(1996-),男,甘肅民勤人,中國科學院地球環境研究所博士研究生,主要研究方向為放射分析化學及環境示蹤.發表論文1篇.
2020-10-28
科技部基礎性工作專項項目(2015FY110800);中國科學院國際合作項目(132B61KYSB20180003);國家自然科學基金資助項目(11875261)
* 責任作者, 研究員, houxl@ieecas.cn
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
今日農業(2021年9期)2021-11-26 07:41:24
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
發明與創新·小學生(2021年3期)2021-03-25 11:48:49
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
汽車工程學報(2017年2期)2017-07-05 08:13:02
中國科技博覽(2016年2期)2016-04-25 20:32:39
小學生導刊(2016年34期)2016-04-11 00:49:44
