基于圖嵌入與拓撲結構信息的蛋白質復合物識別算法*
2021-06-25 09:46:10徐周波劉華東
計算機工程與科學 2021年6期
關鍵詞:監督
徐周波,李 萍,劉華東,李 珍
(桂林電子科技大學廣西可信軟件重點實驗室,廣西 桂林 541004)
1 引言
蛋白質復合物作為大分子組裝體,在細胞穩態、生長和增殖所必需的多種生化活動中發揮著重要作用[1]。蛋白質復合物是生化體制和細胞結構的研究基礎,因此,蛋白質復合物的識別成為近年來的研究熱點。
目前蛋白質復合物識別技術主要分為2類:(1)基于實驗的蛋白質復合物識別技術;(2) 基于計算方法的蛋白質復合物識別技術。基于實驗的蛋白質復合物識別技術主要根據實驗結果識別蛋白質復合物,如免疫共沉淀[2,3]和雙雜交系統[4,5],但其通常耗時較長且需高水平的專業知識作為基礎。為克服基于實驗的蛋白質復合物識別技術的缺點,研究者們提出了多種基于計算方法的蛋白質復合物識別技術。基于計算方法的蛋白質復合物識別技術的基本思想是從蛋白質相互作用PPI(Protein-Protein Interaction) 網絡中識別呈現蛋白質復合物某些典型特性的簇。因此,PPI網絡通常建模為圖的形式,其中圖的節點表示蛋白質,邊表示蛋白質間的相互作用。蛋白質復合物的識別問題可以歸結為一個傳統的圖聚類問題,由此產生的子圖聚類被視為感興趣的蛋白質復合物。Nepusz等[6]提出了聚類算法ClusterONE用于從PPI網絡中發現重疊蛋白質復合物。ClusterONE可有效識別重疊蛋白質復合物,并采用最大匹配率MMR(Maximum Matching Rate)來評估算法的復雜度,該算法精準度和敏感度較低。Liu等[7]提出了一種基于最大團的聚類算法CMC (Clustering-based on Maximal Cliques),利用最大團簇從加權PPI網絡中發現復合群。CMC使用迭代評分的方法為蛋白質對分配權重,可以改善其他蛋白質復合物預測方法的性能,減少隨機噪聲的影響。CMC算法提高了識別蛋白質復合物的精準度,但對小規模復合物檢測能力較差,且敏感度較低。Wang等[8]提出了一種快速分層聚類算法HC-PIN。HC-PIN對假陽性具有魯棒性,并且可以發現低密度的功能模塊。HC-PIN算法雖然也提高了識別蛋白質復合物的精準度,但其同樣存在敏感度低的問題。Wu等[9]提出的COACH算法考慮到蛋白質復合物的拓撲結構,先檢測出核心蛋白質,然后將附屬蛋白質連接到核心蛋白質上。該算法考慮到了蛋白質結構上的特點,一定程度上提高了預測的準確性。Zhao等[10]用不確定圖模型建模PPI網絡,提出了一種基于不確定圖模型的蛋白質復合物算法DCU (Detecting Complex based on Uncertain graph model),改善了COACH算法,進一步提高了預測的準確性。
由于非監督學習算法的隨機特性會在一定程度上影響算法識別結果,因此近年來監督學習算法也逐漸被用于蛋白質復合物的識別。這類算法通過提取樣本特征克服非監督的隨機性,并將特征放入分類器中訓練,最終得出具有一定準確性的分類器。其分類效果的好壞主要依賴于提取的特征能否較好地反映出蛋白質復合物的真實特性。然而,監督學習算法的特征通常都是人為構造的,其準確性和完整性有待考量。
針對傳統算法存在敏感度和F-measure低以及現有監督學習算法中特征構造不完備等不足,近年來許多利用圖嵌入進行蛋白質復合物識別的方法應運而生。圖嵌入的方法將圖轉換為向量的形式進行處理,并且同時保留了圖的局部和全局信息,使得蛋白質復合物的識別更加容易和準確。Xu等[11]提出了一種基于從GO知識庫中學習蛋白質復合物向量的復合物識別算法GANE。該算法利用AANE[12]模型來學習復合物的向量表示,基于此向量構造加權鄰接矩陣并利用團挖掘的算法來進行復合物的識別。Yao等[13]首先將蛋白質以功能不同的標準分組,利用node2vec[14]方法將蛋白質轉換為向量表示,構造相似度矩陣,并利用聚類算法來進行蛋白質復合物的識別。本文提出的graph2vec-SVM與復合物拓撲結構信息相結合的搜索方法,利用graph2vec[15]將圖轉換為向量并結合SVM分類器來進行蛋白質復合物的識別,不僅克服了非監督學習算法的隨機性,還解決了監督學習構造特征不完備等問題,有效彌補了傳統算法和監督學習算法的不足。同時,相較于文獻[14]利用node2vec將復合物中每個節點轉換為向量表示,graph2vec將整個圖轉換為向量表示的做法更加便于計算。通過實驗分析,該算法有較好的敏感度,在準確度和F-measure方面也顯示出良好的性能。
2 相關知識介紹
2.1 graph2vec
PPI網絡通常建模為圖數據模型,圖的節點表示蛋白質,邊表示蛋白質間的相互作用。圖數據模型是一個4元組G=(V,E,W,Lv),其中,V是節點集;E是邊集;W:E→[0,1]是權重分配函數,它給每條邊賦予一個權重;Lv是節點標簽分配函數,它從標簽集中選擇標簽分配給節點。本文以節點度作為圖的標簽,將PPI網絡建模為圖數據模型后利用graph2vec將圖轉換為向量。
graph2vec是由Narayanan等[15]提出的一種圖嵌入(將圖轉換為向量)算法,該算法基于word2vec[16]和doc2vec[17]的思想,將整個圖作為文檔,圖的根子圖作為文檔中的詞,通過訓練淺層神經網絡后最終得到整個圖的向量。其中,根子圖為圖的子樹模式,且子樹中允許出現相同的節點。例如,圖G(圖1a)的最大步長為2的根子圖如圖1b所示。
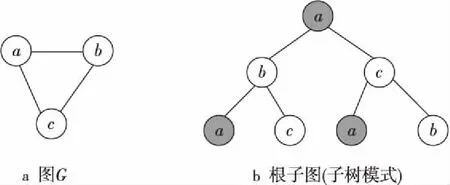
Figure 1 2-rooted subgraph
graph2vec采用skipgram模型來學習圖的向量表示,如圖2所示。給定一個文檔集G={G1,G2,…,Gn}以及從文檔Gq∈G(1≤q≤n)中采樣的詞SG(Gq)={sg1,sg2,…,sgm},skipgram模型通過最大化式(1) 的似然函數得到文檔的向量表示。
(1)
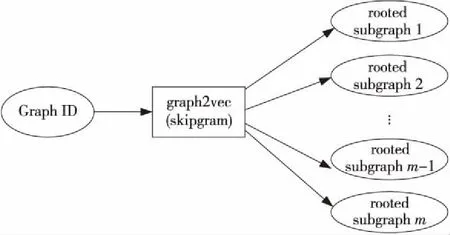
Figure 2 skipgram model
目前現有識別算法通常先將蛋白質復合物建模為圖數據結構,再對其進行特征提取,如圖的密度、節點個數和節點度統計等,并結合機器學習分類器進行蛋白質復合物的識別。由于這些特征是人為構造的,其構造特征的準確性有待考量。graph2vec利用圖本身的特性(每個節點的根子圖),通過skipgram模型訓練后得到的圖的向量表示能夠較準確地保留原圖的信息,且能夠方便地利用機器學習分類器進行后續的蛋白質復合物識別。
2.2 支持向量機
支持向量機SVM(Support Vector Machine)是由Vapnik[18]提出的一種監督學習二分類器。它的基本思想是擬合出一個最大化間隔的劃分超平面,使其能夠具有準確的分類性能。
SVM的劃分超平面可用如式(2)所示的線性方程描述:
wTx+b=0
(2)
其中,x為分類樣本矩陣;w為法向量,決定了超平面的方向;b為位移量,決定了超平面與原點的距離。分類樣本標簽yi為-1或+1,當分類樣本xi能夠被正確分類時,其滿足式(3):
(3)
其中,i表示分類樣本xi在分類樣本矩陣x中的索引,其取值為[0,M],M為分類樣本的總數。式(3)等價于:
yi(wTxi+b)≥1
(4)
滿足yi(wTxi+b)=1的樣本稱為支持向量。2個異類支持向量到劃分超平面的距離之和稱為間隔,即:
(5)
最大化間隔γ即最小化w,求解
s.t.yi(wTxi+b)≥1,i=1,2,…,M
(6)
求解出參數w和b后可得到最大化間隔超平面。
式(6)可用拉格朗日乘子法轉換為對偶問題 ( Dual Problem )的求解,其最后的求解式變為:
(7)

3 基于圖嵌入與拓撲結構信息的蛋白質復合物識別算法
3.1 graph2vec-SVM算法
graph2vec-SVM算法將標準庫中的蛋白質復合物以及隨機生成圖(非蛋白質復合物)用graph2vec技術提取出每個節點的根子圖后,以式 (1) 作為目標函數,利用skipgram模型將圖轉換為向量,轉換后的向量即為SVM分類器的訓練樣本集,然后開始訓練SVM分類器。其主要過程如算法1所示。
算法1graph2vec-SVM
輸入:G={G1,G2,…,Gn},k,N,D,ep,l。
輸出:model。
1.T={};
2.foreachGq∈G
3. randomly generateNsubgraphs fromGqwith the same size asGq,regard them as negative samples and insert them intoT;
4.T=T∪G;
5.vectors=graph2vec(T,k,D,ep,l)
6.model=SVM(vectors,labels);
returnmodel
算法1中,G為蛋白質復合物的集合,k為根子圖的最大步長,N為對每個蛋白質復合物生成隨機子圖的個數,D為向量的維度,ep為graph2vec算法的迭代次數,l為學習率。算法1第1~4行根據每個蛋白質復合物隨機生成N個子圖作為負樣本,并將正負樣本加入訓練集合T中。第5行將訓練集T用graph2vec轉換為向量,第6行將向量和標簽放入SVM分類器中,開始訓練分類器。其中labels為樣本的標簽,正樣本的標簽為+1,負樣本的標簽為-1。
以圖1a為例,設最大步長k為1,則由算法1對圖G提取每個節點的根子圖后,訓練skipgram模型,最終得到圖G的向量表示V(G),如圖3所示。

Figure 3 Steps of graph2vec
3.2 構造候選蛋白質復合物
蛋白質復合物被認為是PPI網絡中的稠密子圖,如何從PPI網絡中劃分出稠密子圖是蛋白質復合物識別的關鍵。本文利用模塊度Q來衡量一個子圖c的稠密程度。子圖c的模塊度Q的定義如式(8)所示:
(8)
其中,Vc為子圖c中的節點集,Ec為子圖c中的邊集。weightin(c)=∑v,u∈VcP(u,v)u,v為子圖c中的節點,P(u,v)為邊e=(u,v)的權重,e∈Ec。weightout(c)=∑v∈Vc,u?VcP(u,v),u為子圖c中的節點,v不為子圖c中的節點,P(u,v)為邊e=(u,v)的權重,e?Ec。δ為模塊校正參數,可用于代表所預測復合物中暫未發現的蛋白質,同時也可用于消除噪聲。Vapnik[18]通過實驗分析,δ取值為PPI網絡平均度的一半時效果最佳。由式(8) 計算得到子圖c的模塊度,若簇邊界內的邊權值總和大于其邊界外的邊權值總和,即:
則稱子圖c為稠密子圖。
由于蛋白質復合物是稠密子圖,在PPI網絡中從度較大的節點開始搜索候選蛋白質復合物,將會更快搜索到稠密子圖,因此本文首先考慮選取節點度大于平均度的節點作為種子節點。由種子節點開始,向外擴散搜索構造子圖,并計算該子圖的模塊度,直至其模塊度達到最大,將其加入候選集中。獲取蛋白質復合物候選集合candidate_set的具體過程如算法2所示。獲取候選集合后,將候選集合中的蛋白質復合物轉換為向量即可用graph2vec-SVM模型進行識別分類。
算法2getcandidate_set
輸入:PPI networkG=(V,E,W,Lv)。
輸出:candidate_set。
1.fornodev∈V/*獲取種子節點集合,種子節點為度大于平均度的節點*/
2.ifdegree ofvmore than average degree ofG,insertvinto the set seed/*由種子節點開始構造候選蛋白質復合物*/
3.fors∈seed
4.c={s};Q(c)=0;
5.Nv(s)//computing the neighbors ofs
6.foreachnoden∈Nv(s)
7.c′=c∪{n};
8. computeQ(c′);//計算子圖模塊度
9.ifQ(c′) ≥Q(c)
10.c=c′;
11. insertcintocandidate_set;
12.returncandidate_set
算法2在執行過程中可能會因為復合物高度重合而造成冗余。本文將候選蛋白質復合物間重合度大于0.7[19]的復合物認為是重合的,重合度計算方法如式(9)所示(即重合度為復合物A和復合物B共有節點個數與復合物A節點個數和復合物B節點個數乘積的比值),并剔除模塊度小的復合物。
OS(A,B)=|A∩B|2/(|A|×|B|)
(9)
去重算法過程如算法3所示。
算法3get finalcandidate_set
輸入:candidate_set。
1.forc∈candidate_set
2.ifSize(c) < 2//丟棄規模小于2的子圖
3. removecfromcandidate_set;
4.forA∈candidate_set
5.forB∈candidate_set
//計算蛋白質復合物間的重合度
6.ifOS(A,B) > 0.7
//保留模塊度大的蛋白質復合物
7.ifQ(A) ≥Q(B)
8. removeBfromcandidate_set
9.elseremoveAfromcandidate_set
3.3 蛋白質復合物的識別
3.1節利用標準庫中的復合物和隨機生成子圖訓練graph2vec-SVM算法并得到具有識別復合物功能的分類器;3.2節利用式(8) 定義的模塊度搜索稠密子圖,去重后得到待識別的候選蛋白質復合物集合;本節利用3.1節中訓練好的graph2vec-SVM算法識別3.2節中去重后得到的候選蛋白質復合物。與算法1相似,在進行蛋白質復合物的識別前,需先利用graph2vec將其轉換為向量,具體過程如算法4所示。
算法4Predict protein complex
輸入:candidate_set,k,D,ep,l。
輸出:predictions。
//用graph2vec將每個候選蛋白質復合物轉換為向量
1.c∈candidate_set
2.vectors=graph2vec(candidate_set,k,D,ep,l);
3.model=graph2vec-SVM(G,k,D,ep,l);
//對候選蛋白質復合物進行識別
4.Predictions=model.predict(vectors);
5.returnpredictions
4 實驗結果及分析
本文將graph2vec-SVM蛋白質復合物識別算法與目前較為經典的4種算法,包括ClusterOne、CMC、HC-PIN和COACH在酵母菌相互作用網絡DIP(Database of Interacting Proteins)[20]上進行比較。蛋白質復合物標準庫采用CYC2008[21]和 MIPS[22]標準庫。2個標準庫分別由408個復合物和428個復合物組成。
4.1 評價指標
本文將所識別的蛋白質復合物與標準庫中的蛋白質復合物進行比較以保證蛋白質復合物識別的質量。識別質量的評價指標主要有精準度(Precision)、敏感度(Sensitivity)和F-measure。 精準度為識別的復合物中真實復合物的數量與識別的復合物總數量的比值;敏感度為識別的復合物中真實復合物的數量與總真實復合物數量的比值;F-measure是精準度和敏感度的調和平均值,其計算方法如式(10)所示:
(10)
Precision=TP/(TP+FP)
(11)
Sensitivity=TP/(FN+TP)
(12)
其中,TP為所識別復合物中與標準庫中復合物相匹配的復合物數量,其匹配程度通過式(9)計算,OS>R的識別復合物被認為是真正的蛋白質復合物,R為匹配程度的閾值,其值通常設置為0.2[23]。TN為識別結果中真實非蛋白質復合物的數量,FN為真實蛋白質復合物被識別為假蛋白質復合物的數量。
4.2 graph2vec參數設置
本文使用標準庫中節點數大于2的蛋白質復合物作為正樣本,負樣本為隨機生成的子圖。利用graph2vec將正樣本和負樣本轉換為向量,參數的設置如表1所示,轉換后的向量即為分類器的訓練集。
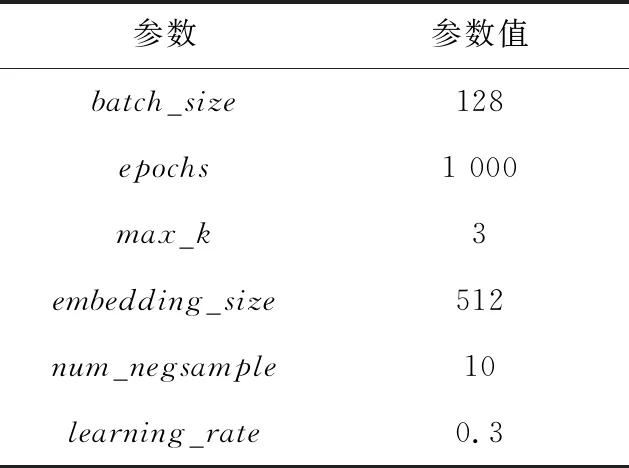
Table 1 Setting of graph2vec parameter
表1中,batch_size為一次訓練所選取的樣本數;epochs為訓練樣本被整體訓練的次數;max_k為根子圖的最大步長;embedding_size為圖轉換為向量的維數,若embedding_size太小會導致圖的信息丟失,從而造成識別算法不能很好地識別出蛋白質復合物,若其太大又會包含冗余的信息,從而影響蛋白質復合物的識別。實驗過程中發現,當embedding_size=512時其能夠較好地表示圖的信息。num_negsamples為噪聲樣本的數量,learning_rate為學習率,最終得到的向量為V(G)=(v1,v2,…,vδ)。
4.3 對比模型的選取
本文在DIP數據集上采用3種機器學習分類器(LR、SVM和XGBoost)進行蛋白質復合物的識別,蛋白質復合物標準庫為MIPS,其結果分別如圖4和表2所示。
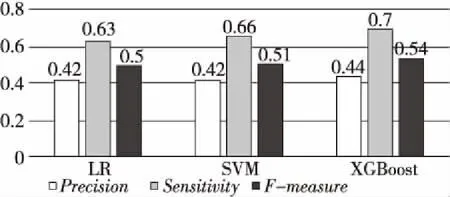
Figure 4 Performance of three classifiers on DIP dataset
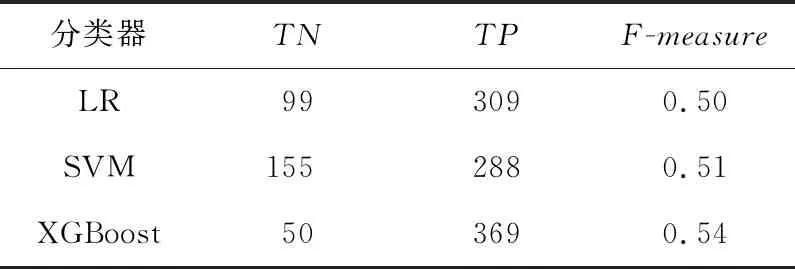
Table 2 Identify results of three classifiers on MIPS standard library
由圖4可知,LR、SVM和XGBoost在3項指標中都有較好的結果,但從表2可看出,LR和XGBoost正確識別蛋白質復合物數量較高,但正確識別非蛋白質復合物的數量極低,而SVM的綜合表現相對較好,所以本文最終選取SVM分類器進行蛋白質復合物的識別。
4.4 與非監督學習算法的對比
graph2vec-SVM與4種非監督學習算法(CMC、COACH、HC-PIN和ClusterOne)在DIP數據集上精準度、敏感度和F-measure的表現如圖5所示,其中蛋白質復合物的標準庫采用的是CYC2008。從圖5可以看出,graph2vec-SVM在3項指標中都取得了良好的效果,在該數據集上的精準度(0.42)有待提高,敏感度(0.66)和F-measure(0.51)均好于其他算法的。
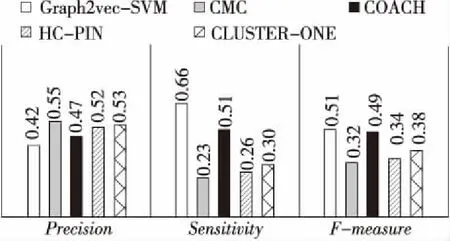
Figure 5 Performance of each algorithm on DIP dataset
為進一步分析實驗結果,將CYC2008標準庫替換為MIPS標準庫后,結果如表3所示。從表3可以看出,graph2vec-SVM識別算法在所有對比算法中識別出正確蛋白質復合物的數量最多,且其F-measure也最高,正確識別非蛋白質復合物的數量比COACH算法次之,但綜合來說graph2vec-SVM識別算法相較于對比算法表現較好。

Table 3 Comparison of algorithms on MIPS standard library
4.5 與監督學習算法的對比
本節將graph2vec-SVM識別算法與3種監督學習算法(SCI-BN,SCI-SVM和RM)在DIP數據集上進行對比。4種算法均采用MIPS標準庫中的蛋白質復合物作為正樣本進行模型訓練。3種監督學習算法參數均參照文獻[23-25]設置。實驗對比結果如表4所示,從表4中可以看出,graph2vec-SVM在DIP數據集上Precision、Sensitivity和F-measure的表現相對其他3種算法都較好。

Table 4 Comparison with supervised algorithms on MIPS standard library
5 結束語
本文針對非監督學習識別算法的隨機特性會影響復合物的識別準確性,以及監督學習識別算法的人為構造特征不完備等缺陷,提出了graph2vec-SVM蛋白質復合物識別算法。該算法利用grah2vec將圖的信息轉換為向量,并進一步采用SVM分類器進行蛋白質復合物的識別,實驗結果表明,該算法與目前流行的監督學習算法與傳統非監督學習算法在敏感度和F-measure上都取得了較好的效果,但由于在生成隨機子圖時存在離散點而導致精準度不高,未來在完善識別算法時我們將著手克服離散點來嘗試提高精準度。
猜你喜歡
新作文·小學低年級版(2022年3期)2022-08-30 07:40:58
公民與法治(2020年15期)2020-09-25 02:57:42
人大建設(2020年4期)2020-09-21 03:39:12
公民與法治(2020年3期)2020-05-30 12:29:40
當代陜西(2019年12期)2019-07-12 09:12:22
消費導刊(2018年10期)2018-08-20 02:57:12
人大建設(2017年2期)2017-07-21 10:59:25
人大建設(2017年9期)2017-02-03 02:53:31
公民與法治(2016年13期)2016-05-17 04:14:35
浙江人大(2014年5期)2014-03-20 16:20:28
