組胺表面分子印跡聚合物的制備及其在酸奶中的應用
2021-06-30 14:56:58李欣培王秀君徐斐曹慧袁敏喻東威劉振民
食品與發酵工業 2021年11期
李欣培,王秀君,徐斐,曹慧,袁敏*,喻東威,劉振民
1(上海理工大學 醫療器械與食品學院,上海食品快速檢測工程技術研究中心,上海,200093) 2(山東黃金礦業股份有限公司,山東 濟南,250014) 3(內蒙古蒙牛乳業集團股份有限公司,內蒙古 呼和浩特,011500) 4(光明乳業股份有限公司乳業研究院,上海,200436)
生物胺(biogenic amine,BA)是一類具有生物活性的低分子質量含氮有機化合物[1],主要由氨基酸脫羧酶作用于氨基酸脫α羧基生成的化合物,其結構可看作生物胺經過轉氨作用,氨分子的1~3個氫原子被烷基或芳基取代[2]。組胺作為最常見的生物胺,屬于單胺類的神經遞質,廣泛存在于各類生物體、富含蛋白質和游離氨基酸的食物中[3],尤其是酸奶、奶酪等發酵乳制品中。適量的組胺在體內有血管舒張的作用,可增加血管通透性、降低血壓,是人體不可缺少的一種化合物[4]。但當人體組胺含量積累到一定的水平或者攝入含量過高時則會導致嚴重的中毒癥狀[5],比如惡心、腹痛、血壓升高、心悸胸悶,甚至呼吸困難[6],癥狀的嚴重程度取決于攝入的組胺含量和個體對組胺中毒的敏感性[7]。
分子印跡技術是人工合成具有特異性識別位點的高分子聚合物的技術[8]。其基本原理是模板分子與功能單體通過共價鍵或非共價鍵的形式組成預聚合物[9],在交聯劑和引發劑的作用下形成均勻的印跡層,之后將印跡聚合物中的模板分子通過洗脫液洗脫,達到特異性吸附的效果。近年來,分子印跡聚合物由于制備簡單[10],穩定性強[11],較好的選擇吸附性[12],可與特定的目標分子結合[13]等優點,在有害物質的分離[14]、去除[15]、化學傳感器[16]以及固相萃取[17-18]等方面實現了廣泛應用。
組胺的分子印跡方法已有研究,GAO等[19]和MAEDEH等[20]采用本體聚合的方法制備了組胺印跡聚合物,將模板分子與功能單體在引發劑和交聯劑的作用下形成固體塊狀、硬度較大的聚合物,但后期還需要進行機械研磨,研磨過程中易破壞印跡空腔,且由于模板分子包埋較深,后續洗脫過程耗時較長。表面分子印跡方法是指將分子識別位點設計在載體表面或接近表面的地方[21],與傳統的本體聚合相比具有吸附更快、吸附容量更高的優點。本文基于分子印跡技術,制備了一種吸附速率快,吸附容量高的印跡聚合物,在后續的檢測研究中起到快速高效的吸附效果。本文采用以乙烯基修飾的SiO2為載體,利用表面聚合的方法制備組胺的表面分子印跡聚合物(molecularly imprinted polymer,MIP),同時對其吸附性能進行了研究。
1 材料與方法
1.1 試劑與儀器
組胺(分析純)、酪胺(優級純)、二甲基丙烯酸乙二醇酯(純度98%)、正硅酸乙酯(優級純)、3-(三甲氧基甲硅基)甲基丙烯酸丙酯(純度≥97%),美國Sigma-Aldrich公司;甲基丙烯酸(色譜純),阿拉丁工業有限公司;無水乙醇(分析純)、氨水(28%,分析純)、甲醇(色譜純)、乙酸(分析純)、甲苯(色譜純)、組氨酸(生物試劑),國藥集團化學試劑有限公司;衣康酸(分析純)、色胺(純度98%),上海麥克林生化科技有限公司;偶氮二異丁腈(分析純),天津市光復精細化工研究所。
TU-1901雙光束紫外可見分光光度計,北京普析通用儀器有限公司;SP200-2T多通道磁力攪拌器,杭州米歐儀器有限公司;KQ-400KDE高功率數控超聲波清洗器,昆山市超聲儀器有限公司;Sorvall ST 8R高速冷凍離心機,賽默飛世爾科技有限公司;DF-101S集熱式恒溫加熱磁力攪拌器,鞏義市予華儀器有限責任公司;Waterse 2695高效液相色譜儀、Hypersil ODS色譜柱(150.0 mm×4.6 mm,5 μm),美國沃特世公司。
1.2 印跡聚合物的合成
1.2.1 印跡載體SiO2-MPS的制備
向120 mL無水乙醇中攪拌加入50 mL超純水、10 mL的正硅酸乙酯(tetraethyl orthosilicate,TEOS)和6 mL氨水(28%),在室溫下旋勻8 h至溶液呈乳白色,得到SiO2微球懸濁液,離心并用乙醇清洗3遍后干燥備用。
稱取1 g干燥的SiO2微球,加入3 mL 3-(三甲氧基甲硅基)甲基丙烯酸丙酯(3-(trimethoxysilyl)propyl methacrylate,MPS),并將其分散于100 mL甲苯中,90 ℃下攪拌24 h,將MPS修飾在硅球表面。反應結束后使用乙醇洗掉未結合的甲苯,干燥備用。
1.2.2 組胺表面分子印跡聚合物的制備
將11.1 mg的模板分子組胺與84.8 μL甲基丙烯酸(methacrylic acid,MAA)溶于15 mL無水乙醇溶液中,超聲5 min混勻后在4 ℃下攪拌1 h進行預聚合。在上述溶液中加入200 mg的載體SiO2-MPS、754.4 μL交聯劑二甲基丙烯酸乙二醇酯(ethylene glycol dimethacrylate,EGDMA)和10 mg引發劑偶氮二異丁腈(azodiisobutyronitrile,AIBN),氮吹20 min后密封燒瓶,在60 ℃條件下聚合20 h。聚合完成后,取出聚合物8 000 r/min (12 000×g)離心棄去上清液,使用無水乙醇洗掉未結合的試劑。取30 mL甲醇/乙酸(9∶1,體積比)作為洗脫溶液,磁力攪拌1 h后超聲振蕩30 min,7 000 r/min離心5 min后去除上清液,重復上述步驟5~6次,直至離心后的上清液中不含有組胺。非印跡聚合物(non imprinted polymer,NIP)除不使用組胺外,制備流程同上。
1.3 組胺分子印跡聚合物吸附平衡濃度和吸附時間的優化
稱取等量10 mg的MIP,加入10 mL不同濃度的組胺溶液,混勻后吸附30 min,離心取上清液,測定210 nm處的吸光度值,根據吸附前后溶液中含有的組胺的濃度變化,采用差減法計算MIP的吸附容量Q[22],如公式(1)所示:
(1)
式中:c0,組胺溶液的初始濃度,μmol/L;c1,吸附后溶液中組胺的濃度,μmol/L;V,吸附時的溶液體積,L;M,組胺的相對分子質量,g/moL;m,印跡聚合物的質量,g。
稱取等量10 mg的MIP和NIP,加入10 mL 的0.5 mmol/L組胺溶液,混勻后分別吸附10、20、30、60、90、120 min后離心取上清液,測定210 nm處的吸光度值。分子印跡中通常使用印跡因子(imprinting factor,IF)來表示印跡效果[22],按公式(2)進行計算:
(2)
式中:QMIP,MIP的吸附容量,mg/g;QNIP,NIP的吸附容量,mg/g。
1.4 組胺分子印跡聚合物特異性吸附
由于不同的基團其紫外吸收最大波長也不同,因此分別測量組氨酸、苯乙胺、酪胺3種組胺結構類似物的最大吸收波長后,確定其各自的標準曲線。分別配制相同濃度的組氨酸溶液、酪胺溶液和苯乙胺溶液各10 mL,加入等量的10 mg的MIP,混勻后吸附同樣的時間,離心取上清液,測定吸附前后210 nm處紫外吸光度值的變化,計算吸附容量。
1.5 對酸奶中組胺的吸附
在當地超市選購酸奶,利用所制備的印跡聚合物進行組胺的加標回收檢測。取4 mL酸奶樣品,加入1 mL不同濃度的組胺標準液,靜置30 min,旋勻使其加標濃度分別為200、300、500 μmol/L,然后加入20 mL 0.1%的三氯乙酸和5 mL 2%的乙酸鉛振蕩5 min,7 000 r/min離心后抽濾上清液。每一個加標濃度的酸奶樣品分別制備3批樣品以進行平行實驗。將10 mg的印跡聚合物與10 mL加標的實際樣品進行10 min的旋勻結合,吸附后離心取上清液進行紫外吸收光譜測定,并計算其吸附容量。同時,利用高效液相色譜儀進行驗證測定。
色譜條件:A相為90%乙腈和10%的水相(0.77 g乙酸銨溶于10 mL乙酸溶液后定容至1 L),B相為90%水相和10%的乙腈;梯度洗脫程序表參考GB 5009.208—2016《食品安全國家標準 食品中生物胺的測定》。
2 結果與分析
2.1 功能單體種類及比例的篩選
模板分子與功能單體的比例對印跡聚合物的吸附性能有比較重要的影響。功能單體過多時,較大的空間位阻可能會導致模板分子在后續洗脫過程中難以洗脫干凈;功能單體比例較少時,制備的印跡聚合物對模板分子的非特異性吸附要多于特異性吸附,可能會出現假陽性的結果。
由于組胺分子含有的氨基和含氮雜環易與單體形成氫鍵結構[23],因此可以通過紫外分光光度計測定和衣康酸(itaconic acid,ITA)2種功能單體與組胺的結合情況。分別配制1 mmol/L的組胺-乙醇溶液、5 mmol/L的MAA-乙醇溶液和5 mmol/L的ITA-乙醇溶液,以MAA為例,于7個燒瓶中分別加入100 μL的組胺-乙醇溶液,然后加入不同體積的MAA-乙醇溶液,分別配制成7種不同組胺與單體比例的10 mL混合溶液,體積比分別為1∶2、1∶4、1∶6、1∶8、1∶10、1∶12、1∶15,超聲混勻后在4 ℃培養箱中反應1 h。根據吸光度的加和性,若2種物質不發生反應,則其混合溶液的吸光度理論上為兩者單獨吸光度的加和;兩者發生反應后吸光度會發生變化,并隨著反應結合能力的增強,其變化越明顯[24]。根據此原理測定了預聚合溶液在190~240 nm的紫外吸收光譜,并對結合前后的最大吸收峰值進行差值比較,結果如圖1所示。
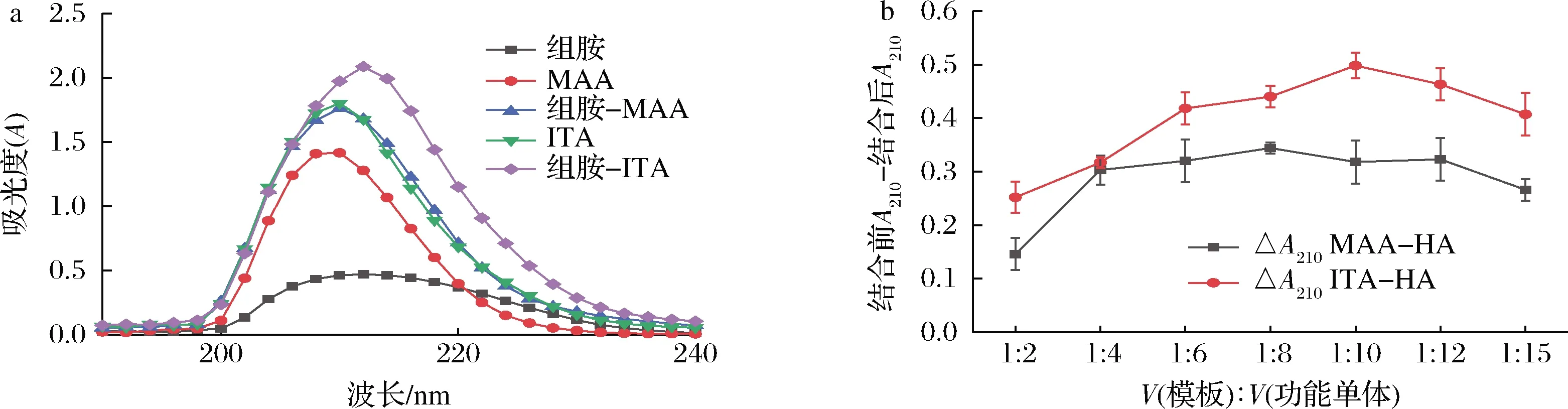
a-紫外吸收掃描曲線;b-紫外吸光值變化圖1 不同功能單體與模板預聚合后的紫外吸收掃描曲線和210 nm紫外吸光值的變化Fig.1 UV absorption scanning curves and the absorbance changes at 210 nm of the prepolymer with different functional monomers and template
由圖1-a所示,當以MAA為功能單體時,組胺與MAA預結合后,其吸光度差值最大的比例為1∶8;而以ITA為功能單體時,其最佳結合比例為1∶10。因此,在交聯劑和引發劑固定的情況下,分別比較了不同功能單體比例(1∶4、1∶6、1∶8、1∶10和1∶12)下MAA、ITA所制備的印跡聚合物的吸附容量,進而確定單體以及單體與模板的比例。結果如圖2所示,在模板與單體的比例為1∶10時,MAA和ITA對組胺的吸附容量均高于1∶8比例時的吸附容量,其中MAA對組胺的吸附容量達到1.9 mg/g,遠高于ITA的吸附容量(0.55 mg/g)。綜合以上結果,ITA與組胺分子結合能力強,應當歸因于ITA分子中的2個羧基容易與組胺結合,但其分子結構要比MAA大,且在分子印跡過程中,產生的印跡空腔要比MAA少,因此考慮MAA作為最終的功能單體,單體與模板比例采用1∶10。
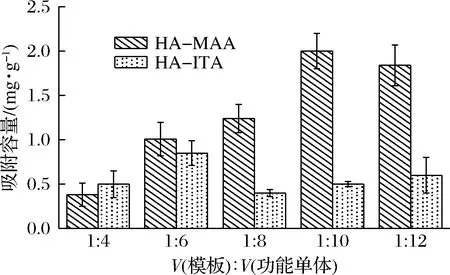
圖2 MAA和ITA制備的表面印跡聚合物對組胺的吸附容量Fig.2 Histamine adsorption of surface imprinted polymers prepared by MAA and ITA
2.2 交聯劑含量優化
在聚合過程中,交聯劑的作用是將預結合后的單體與載體之間形成交聯,形成具有一定結構的高分子聚合物,在模板分子去除后形成與其形狀互補的孔穴。交聯劑的種類和數量會直接影響MIP的吸附性能,交聯劑過少會導致聚合物無法支撐孔穴結構,過多則會導致聚合物交聯成塊,影響模板的洗脫和后續檢測時對模板的識別與吸附。
本文比較了不同含量的交聯劑對印跡聚合物吸附性能的影響。將11.1 mg的模板分子組胺與84.8 μL的MAA溶于15 mL的無水乙醇溶液中,超聲混勻后4 ℃下攪拌1 h。在固定了組胺和MAA含量的情況下,分別加入94.3、188.6、377.2、565.8、754.4、943.0 μL的EGDMA,配制6種模板與交聯劑不同含量比例的印跡聚合物,比例分別為1∶5、1∶10、1∶20、1∶30、1∶40、1∶50。
印跡聚合物的吸附容量結果如圖3所示,隨著交聯劑的用量從94.3 μL增加到754.4 μL,模板與交聯劑EGDMA的體積比從1∶5增加到1∶40,相應制備的印跡聚合物的吸附容量也從2.3 mg/g增加到5.99 mg/g。而當交聯劑含量增加至943.0 μL時,模板與交聯劑體積比達到1∶50,吸附容量僅有2 mg/g,此時的印跡聚合物中含有的交聯劑過多,形成的印跡聚合物反而不利于模板分子的識別。由于2.1小節中優化的模板與功能單體的最佳比例為1∶10,后續實驗中將模板、功能單體和交聯劑的比例確定為1∶10∶40。

圖3 不同交聯劑比例下MIP的吸附容量Fig.3 Adsorption capacity of MIP with different crosslinking agent ratio
2.3 印跡聚合物MIP的結構表征
利用透射電子顯微鏡對聚合物的形狀與粒徑進行分析,由圖4-a可以看出,所制備的SiO2分散均勻,形態均一呈圓球型,直徑在300~400 nm。在印跡聚合物(b)的透射電鏡顯微圖中,可看到在SiO2微球表面覆蓋了一層厚度約40~50 nm的印跡層,表明印跡聚合物已在微球表面制備成功。

圖4 硅球載體(a)和帶有印跡聚合物的硅球(b)的透射電鏡顯微圖Fig.4 Transmission electron microscope of silica ball carrier (a) and silica ball with imprinted polymer (b)
同時利用紅外光譜儀,對載體和聚合物進行了紅外光譜分析。圖5展示了SiO2-MPS、未洗脫模板的MIP-HA和洗脫模板后的MIP的紅外光譜圖。
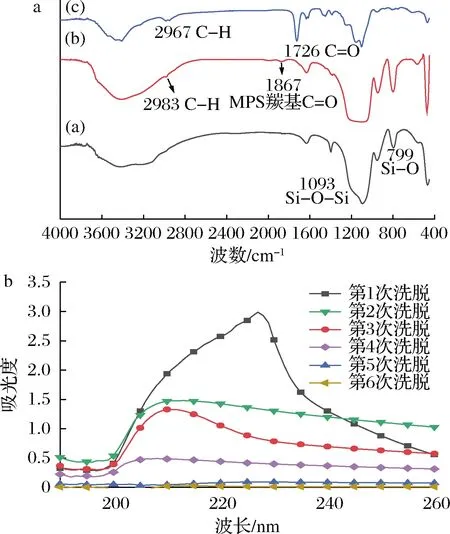
a-MIP;b-MIP-HA;c-SiO2-MPS圖5 SiO2-MPS、未洗脫模板的MIP-HA和洗脫模板后的MIP的紅外光譜圖Fig.5 Infrared spectra of SiO2 MPs, MIP-HA without eluting template and MIP after eluting template

2.4 組胺分子印跡聚合物的吸附特性
為了探究印跡聚合物對組胺分子的吸附效果,在0.1~0.6 mmol/L的組胺加標濃度范圍內以吸附容量作為指標,進行了吸附試驗。如圖6所示,在0.1~0.5 mmol/L的濃度范圍內隨著組胺濃度的增加,印跡聚合物的吸附容量也隨之增加。組胺濃度為500 μmoL/L時,MIP對模板分子的吸附容量達到最大值,為13.1 mg/g。
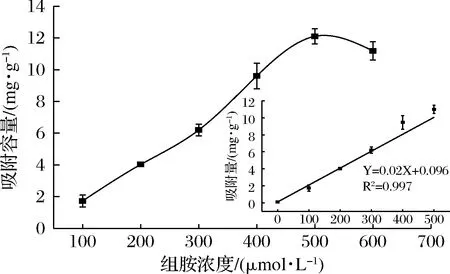
圖6 組胺印跡聚合物的吸附等溫線Fig.6 Adsorption isotherms of histamine imprinted polymers
2.5 組胺分子印跡聚合物的動力學分析
吸附平衡時間反應了印跡聚合物的吸附效率。相同濃度下的吸附時間變化如圖7所示。在最初10 min內,組胺的吸附容量迅速增加,達到一個幾乎隨時間延長而保持不變的最大值,說明印跡聚合物的吸附過程已達到平衡。而且MIP的最大吸附容量為13.1 mg/g時,NIP的吸附容量僅有3.3 mg/g,MIP的吸附能力約是NIP的4倍,因此MIP對組胺分子的親和性和特異選擇性要比NIP好。并且與以往研究中本體聚合方法[25]所制備的印跡聚合物相比,本研究制備的聚合物吸附速率快,到達吸附平衡的時間更短。

圖7 組胺印跡聚合物的動力學吸附曲線Fig.7 Kinetic adsorption curve of histamine imprinted polymer
2.6 印跡聚合物的特異性分析
選取組氨酸、酪胺和苯乙胺這3種組胺的結構類似物,將所制備的印跡聚合物與之反應,進行吸附容量測定,結果如圖8所示。在500 μmol/L加標濃度下,所制備的印跡聚合物對組胺有很好的吸附性,對結構類似物的吸附性較低。與組胺的前體物質組氨酸相比,所制備的印跡聚合物對組胺的吸附容量是組氨酸吸附容量的13倍。
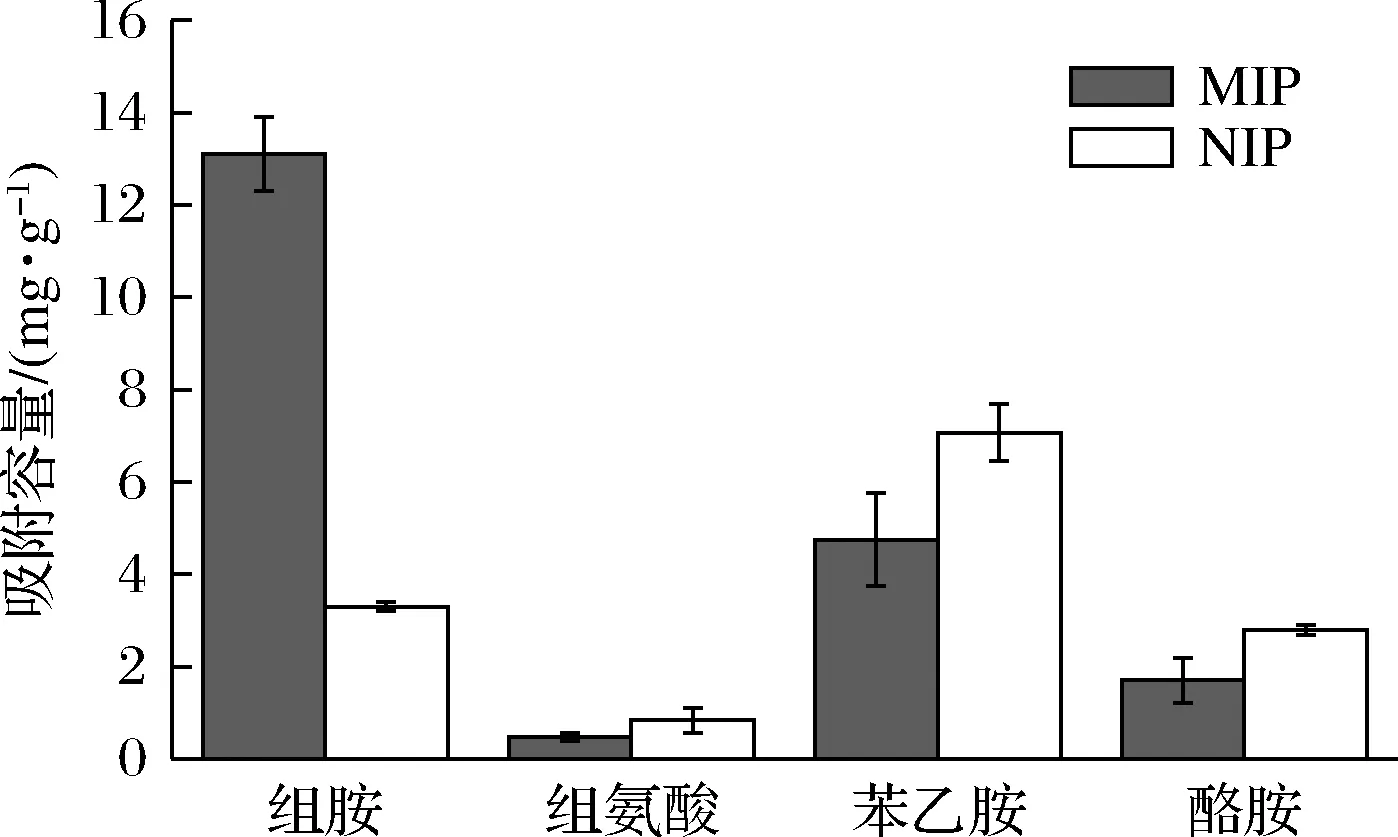
圖8 印跡聚合物對組胺及其結構類似物的吸附能力Fig.8 Adsorption capacity of imprinted polymers for histamine and its analogues
2.7 與以往研究的對比
在以往組胺分子印跡聚合物的研究當中,通常采用本體聚合的方法,所需吸附時間為1~4 h,無法達到快速檢測的目的。本研究方法可在10 min內快速達到吸附平衡且吸附容量高于其他已公開的印跡聚合物制備方法(表1)。

表1 研究方法對比Table 1 Comparison of research methods
2.8 對酸奶中組胺的吸附
以1,7-二氨基庚烷為內標,組胺的液相色譜圖如圖9所示,以組胺的峰面積與內標峰面積為比值,繪制標準曲線。通過對酸奶樣品進行200、300和500 μmol/L這3個水平的組胺加標,使用高效液相色譜測定了該MIP在實際樣品中的吸附性能,如表2所示,當組胺濃度為500 μmol/L時,MIP的吸附容量達到8.35 mg/g,并且通過與組胺標準溶液中的吸附性能進行比較,結果顯示在酸奶樣品中,該印跡聚合物仍具有良好的吸附性能,10 mg的印跡聚合物去除率達到20%,后續可通過增加聚合物的使用量來進一步增大去除率。
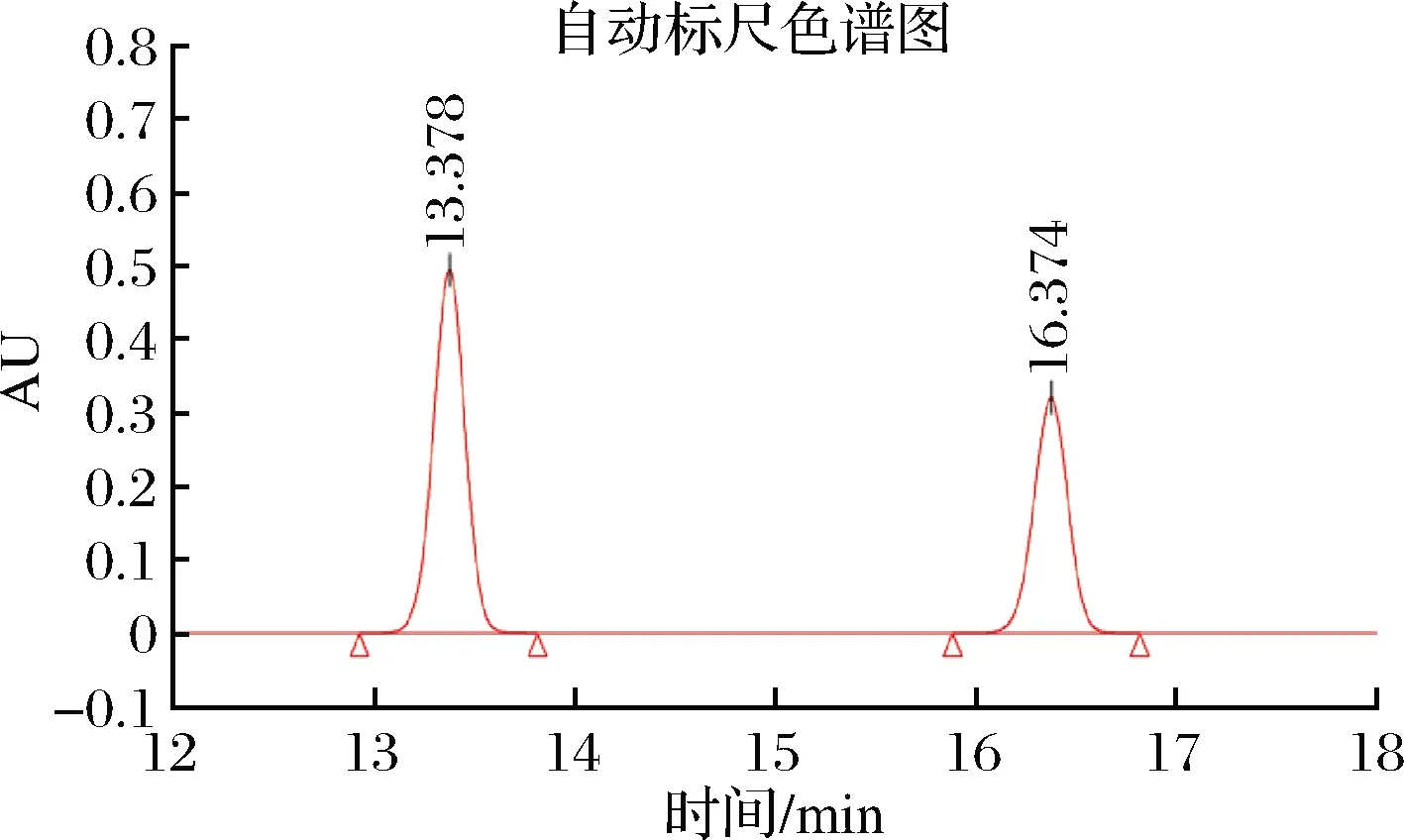
圖9 組胺和內標的高效液相色譜圖Fig.9 High performance liquid chromatography of histamine and internal standard
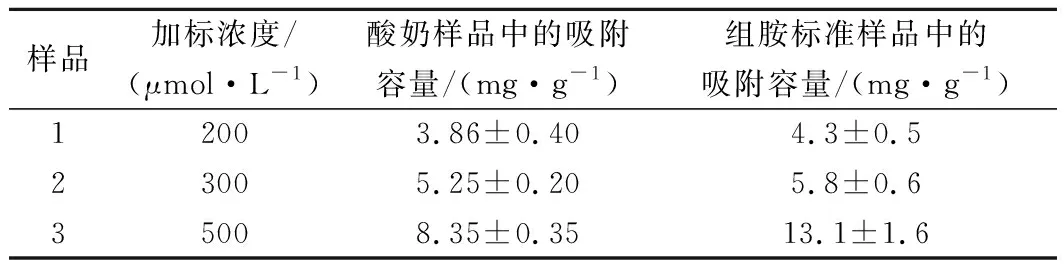
表2 實際樣品的吸附測定Table 2 Adsorption determination of actual samples
3 結論
以組胺為模板分子,甲基丙烯酸為功能單體,無水乙醇為溶劑,在雙鍵修飾的二氧化硅表面上制備了具有特異性的表面分子印跡聚合物,并通過紅外光譜和透射電鏡進行表征,驗證了表面印跡聚合物的合成。與傳統的本體聚合方法相比,該表面印跡聚合物對組胺有更大的吸附容量(13.1±1.6) mg/g,吸附速度更快,可在10 min內達到吸附平衡。該表面印跡聚合物也展現出了良好的選擇性,其吸附容量是結構類似物組氨酸的13倍,該聚合物也被成功應用于實際樣品酸奶中組胺的吸附。
