畢赤酵母醇氧化酶2啟動子上游調控序列的隨機突變研究
2021-06-30 00:59:20胡詩藝熊紫瑩周化嵐張建國
工業微生物 2021年3期
劉 冰,胡詩藝,熊紫瑩,韋 雪,周化嵐,張建國
上海理工大學 醫療器械與食品學院 食品科學與工程研究所,上海 200093
畢赤酵母(Komagataellaphaffii,曾命名Pichiapastoris) 是能夠利用甲醇作為唯一碳源生長的甲基營養型微生物,是目前一種重要的表達外源蛋白的工業微生物[1]。其過氧化酶體中的醇氧化酶1(AOX1)和醇氧化酶2(AOX2)將甲醇氧化為甲醛。醇氧化酶對甲醇和氧氣的親和力比較低[2,3],因此,需要大量的醇氧化酶來代謝甲醇。但是PAOX1和PAOX2都受甲醇嚴格調控,調控機制相同[4,5]。畢赤酵母在含有葡萄糖、乙醇、甘油等碳源的培養基中生長時,PAOX1和PAOX2都被抑制,細胞中無法檢測到AOX1和AOX2的mRNA[2,6-7]。PAOX1和PAOX2在甲醇為唯一碳源的培養基中表達。由于醇氧化酶2啟動子(PAOX2)比醇氧化酶1啟動子(PAOX1)弱,導致AOX2的含量只占總醇氧化酶量的5%[6,7]。AOX1的基因序列(aox1)和AOX2的基因序列(aox2)高度同源,但它們的啟動子區域的DNA序列沒有同源性。醇氧化酶啟動子強度的差異是由醇氧化酶啟動子上游轉錄調控序列控制,若將aox2置于PAOX1控制下,重組菌株具有AOX1相似的活力。相反,若將aox1置于PAOX2控制下,AOX1的表達量顯著下降[6]。目前PAOX1因其調控嚴格,表達能力強等原因而被廣泛研究[8-14],但是依然沒有明確其調控序列。而PAOX2的上游調控序列相關信息更少[15,16]。因此,突變PAOX2這樣的弱啟動子會比強啟動子PAOX1更容易實現外源蛋白表達量的升高[17]。而且,研究PAOX2的上游調控序列較易得到相關調控序列,有利于闡明醇氧化酶啟動子的調控序列。OHI 等[15]對畢赤酵母PAOX2上游調控序列進行敲除和插入序列,鑒定出PAOX2的一個上游激活序列(UAS)和兩個上游抑制序列(URS)。其中UAS與PAOX1的UAS相似。另外,戴秀玉[18]等從aox1基因缺陷菌株Muts中篩選得到Mut+表型突變體,經過測序后發現PAOX2的突變增強了aox2基因的轉錄,因此可以通過改變PAOX2的上游調控區域來改善aox1基因缺陷菌株利用甲醇的能力,使其在基因工程藥物表達及產業化方面發揮重要作用。
本研究以GH11家族的黑曲霉木聚糖酶(Xylanase,xynB)[19,20]作為外源蛋白的模型,對PAOX2上游調控序列進行隨機突變,構建了PAOX2突變型畢赤酵母文庫,根據表達木聚糖酶酶活的差別,篩選突變菌株并對其PAOX2進行測序,最后分析突變位置對PAOX2表達能力的影響。
1 材料與方法
1.1 試劑和儀器
本研究實驗的生化試劑盒、主要儀器見表1。

表1 試劑和儀器
1.2 載體及菌株
質粒pPIC9K、pPIC9K-PAOX2-xynB由實驗室保藏;大腸桿菌Top 10購自TaKaRa公司;畢赤酵母 GS115菌株由實驗室保藏。
1.3 培養基與溶液的配制
Luria-Bertani(LB)培養基:稱取1 g蛋白胨、0.5 g酵母粉、1 g氯化鈉,溶于100 mL去離子水水中,調整pH至7.0。如制作平板,則加入1.5 g瓊脂粉。121 ℃高壓滅菌15 min。
Yeast Extract Peptone Dextrose (YPD)培養基:稱取1 g 酵母粉、2 g蛋白胨、2 g葡萄糖,溶于100 mL去離子水中。如需固體培養基,則加入1.5 g瓊脂粉。115 ℃高壓滅菌20 min。配置好的YPD培養基于室溫存放,平板置于4 ℃儲存。
Minimal Dextrose(MD)培養基:稱取1.34 g YNB、2 g葡萄糖、1.5 g瓊脂粉(配制平板時添加)溶解于100 mL去離子水中,115 ℃高壓滅菌20 min。使用前加入200 μL濃度為4×10-5g/L的無菌生物素。配置好的培養基于室溫存放,平板置于4 ℃冰箱存放。
磷酸鹽緩沖液 (PBS):稱取8 g氯化鈉、0.2 g氯化鉀、3.63 g十二水合磷酸氫二鈉、0.24 g磷酸二氫鉀,溶解于900 mL去離子水中,用鹽酸調整pH,室溫存放。
Buffered Glycerol-complex (BMGY)培養基:稱取酵母粉1 g、蛋白胨2 g溶解于70 mL 去離子水中,115 ℃高壓滅菌20 min。量取10 mL 1 mol/L PBS緩沖液(pH視情況調整),115 ℃高壓滅菌20 min。稱取1.34 g YNB溶解于10 mL去離子水中,115 ℃高壓滅菌20 min。稱取1.26 g甘油溶解于9 mL 去離子水中,115 ℃高壓滅菌20 min。上述四種滅菌試劑于室溫存放,使用前將其混合到一起并加入200 μL無菌的 4×10-5g/L 生物素,充分混勻。
檸檬酸-磷酸緩沖溶液:稱取2.9244 g磷酸氫二鈉、2.04 g檸檬酸溶解于200 mL去離子水中,充分溶解混勻后則制成pH為5.0,濃度為50 mmol/L的檸檬酸-磷酸緩沖溶液,室溫存放。
木聚糖溶液:將1 g木聚糖溶解于100 mL pH為5.0,濃度為50 mmol/L的檸檬酸-磷酸緩沖溶液中,充分混勻制成1% 的木聚糖溶液。由于木聚糖溶液穩定性不高,應盡量在使用前配制,室溫存放。
木糖溶液:準確稱量分析純的無水木糖100 mg(預先在105 ℃干燥至恒重),加入少量去離子水溶解,定容至100 mL,充分搖勻后即獲得濃度為1 mg/mL的木糖溶液。
DNS:將1.071 g/mL的苯酚于50 ℃充分融化,稱取91.1 g酒石酸鉀鈉、3.15 g 3,5-二硝基水楊酸溶解于去離子水中,加入10.5 g氫氧化鈉和2.33 mL融化的苯酚,再加入2.5 g偏重亞硫酸鈉,將溶液定容至500 mL。配制好的溶液應存放于4 ℃,并于配好后7 d使用,有效期為6個月。
1.4 PAOX2序列的隨機突變
以重組質粒pPIC9K-PAOX2-xynB為模板,TB-AOX2-F/R (TB-AOX2-F:5′-tcgagatctgagctcgaattctttt-3′;TB-AOX2-R:5′-tctcatcgtttggatccttttctcagt-3′)為引物,利用Diversify?隨機突變試劑對PAOX2進行PCR。PCR反應程序為94 ℃預變性30 s;94 ℃變性30 s,54 ℃退火延伸96 s,共循環25次;最后再54℃延伸100 s,4℃冷卻。將擴增后的產物經過2%瓊脂糖凝膠電泳分析,并利用PCR清潔試劑盒對隨機擴增產物進行純化。
重組質粒pPIC9K-PAOX2-xynB經限制性內切酶BamH 1和EcoR 1消化后,回收雙酶切位點BamH 1之間的片段(593 bp)以及酶切位點BamH 1和EcoR 1之間的片段(8 295 bp),然后利用T4連接酶對以上兩個片段進行連接(16 ℃連接4 h)。利用PCR清潔試劑盒對T4酶連接后的產物進行純化,然后利用In-fusion技術對純化后的連接產物與突變后的PAOX2片段進行連接。然后將此連接產物轉化至大腸桿菌TOP 10感受態中,并在含有氨芐的抗性平板上篩選。將平板置于37 ℃培養箱中12 h~16 h,待平板上長出單菌落之后挑取單菌落,以TB-AOX2-F/R為引物進行菌落PCR驗證并測序,測序由生工生物工程(上海)股份有限公司完成。
1.5 PAOX2突變型畢赤酵母的構建和培養
PAOX2突變后的重組質粒經Sal1酶切線性化后,電轉至畢赤酵母 GS115感受態細胞后涂MD平板培養基。MD平板培養基經30℃培養至長出單菌落,分別挑取突變型畢赤酵母單菌落接種于50 mL YPD培養基中,在30 ℃、200 r/min搖床中培養24 h。將突變型畢赤酵母種子液按4%的接種量接種到20 mL BMGY培養基中,在30 ℃、200 r/min搖床中培養48 h,使其培養基中的甘油耗盡,此時添加1%的甲醇進行誘導,之后每隔24 h補加1%甲醇,共誘導96 h。在每次補加甲醇之前將發酵液充分吸打混勻,然后取樣。最后于4 ℃,12 000 g條件下離心發酵液10 min。收集上清,測定樣品中的木聚糖酶酶活。每株菌株的誘導設計三個平行樣。
1.6 木聚糖酶酶活力定義及測定方法
向2 mL 1% 的木聚糖溶液中加入20 μL 發酵液離心上清(空白對照組用去離子水代替),迅速吸打混勻后,50 ℃水浴5 min。迅速加入2 mL DNS溶液和1 mL 水后吸打混勻,100 ℃水浴 10 min 顯色。然后迅速取出加入去離子水定容至20 mL。充分混勻后,利用酶標儀在540 nm處測定光吸收值,最后計算每個樣品的酶活。每個反應測定三個平行樣。木聚糖酶酶活力單位的定義為:在標準條件下,每分鐘水解1%木聚糖形成1 μmol/L木糖(還原糖)所需酶量為1個酶活力單位[21]。
2 結果與分析
2.1 PAOX2的隨機突變及PAOX2突變型畢赤酵母的構建
通過改變PCR反應中Mn2+和dGTP的量來控制隨機突變水平,對PAOX2進行隨機突變。擴增后的產物經2% 瓊脂糖凝膠電泳分析顯示突變后的PAOX2大小約為1 550 bp(圖1-a)。重組質粒pPIC9K-PAOX2-xynB被BamH 1酶和EcoR 1消化后,經1% 瓊脂糖凝膠電泳分析顯示酶切產物共有三個片段,大小分別約為 8 300 bp,1 500 bp和600 bp(圖1-b)。將雙酶切位點BamH 1之間的片段(593 bp)以及酶切位點BamH 1和EcoR 1之間的片段(8 295 bp)進行T4連接,連接產物經1%瓊脂糖凝膠電泳分析顯示其大小約為8 900 bp(圖1-c)。T4連接產物與突變后的PAOX2片段經In-fusion技術連接后轉化大腸桿菌,隨機挑取9株大腸桿菌進行菌落PCR驗證,驗證結果如圖1-d所示,PCR產物經2%瓊脂糖凝膠電泳分析顯示其大小約為1 600 bp。測序顯示其中4株菌株的PAOX2已經發生突變,表示約有50%的片段已發生突變,說明成功構建了pPIC9K-PAOX2-xynB啟動子突變文庫。

圖1 PAOX2突變質粒的構建
將啟動子突變型重組質粒酶切后電轉入畢赤酵母中,即獲得了PAOX2突變型畢赤酵母文庫(圖2)。
圖2 PAOX2突變型畢赤酵母
2.2 PAOX2突變型畢赤酵母的篩選
以野生型PAOX2為啟動子的重組畢赤酵母GS115-pPIC9K-PAOX2-xynB為對照組,挑取了850株PAOX2啟動子突變型畢赤酵母進行了培養。圖3展示了850株PAOX2突變型畢赤酵母表達木聚糖酶的活力,按照活力的高低分為6組。對照組酶活的0%~20%、20%~40%、40%~60%、60%~80%、80%~100%、100%~120%區間分別有207、58、43、44、260、238株突變型酵母菌。約有59%的突變菌株表達的木聚糖酶活力為對照組菌株的80%~120%,約有24%的突變菌株表達的木聚糖酶活力為對照組菌株的0%~20%,最后,約有17%的突變型菌株表達的木聚糖酶活力為對照組菌株的20%~80%,此區間內的酶活顯著低于或稍微低于對照組菌株的酶活,這可能是由于突變造成了PAOX2表達木聚糖酶的能力下降。圖4展示了20%~40%、40%~60%和60%~80%三個組別的菌濃度,他們的OD600分別為29.45±2.24、30.02±4.22和28.79±3.44,菌濃度基本相同,說明此次PAOX2隨機突變對菌的生長沒有明顯影響。
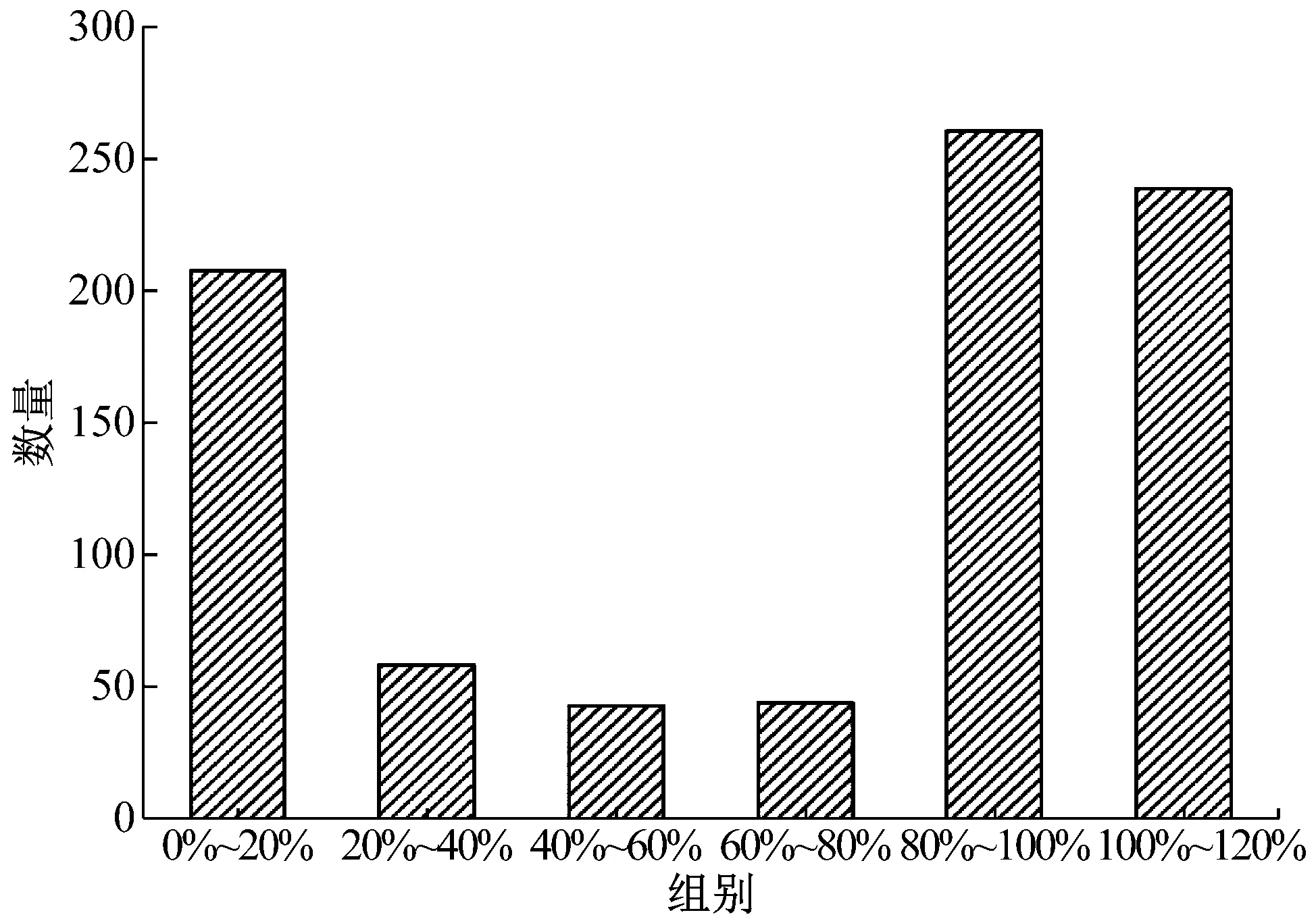
圖3 不同表達量的菌株數量
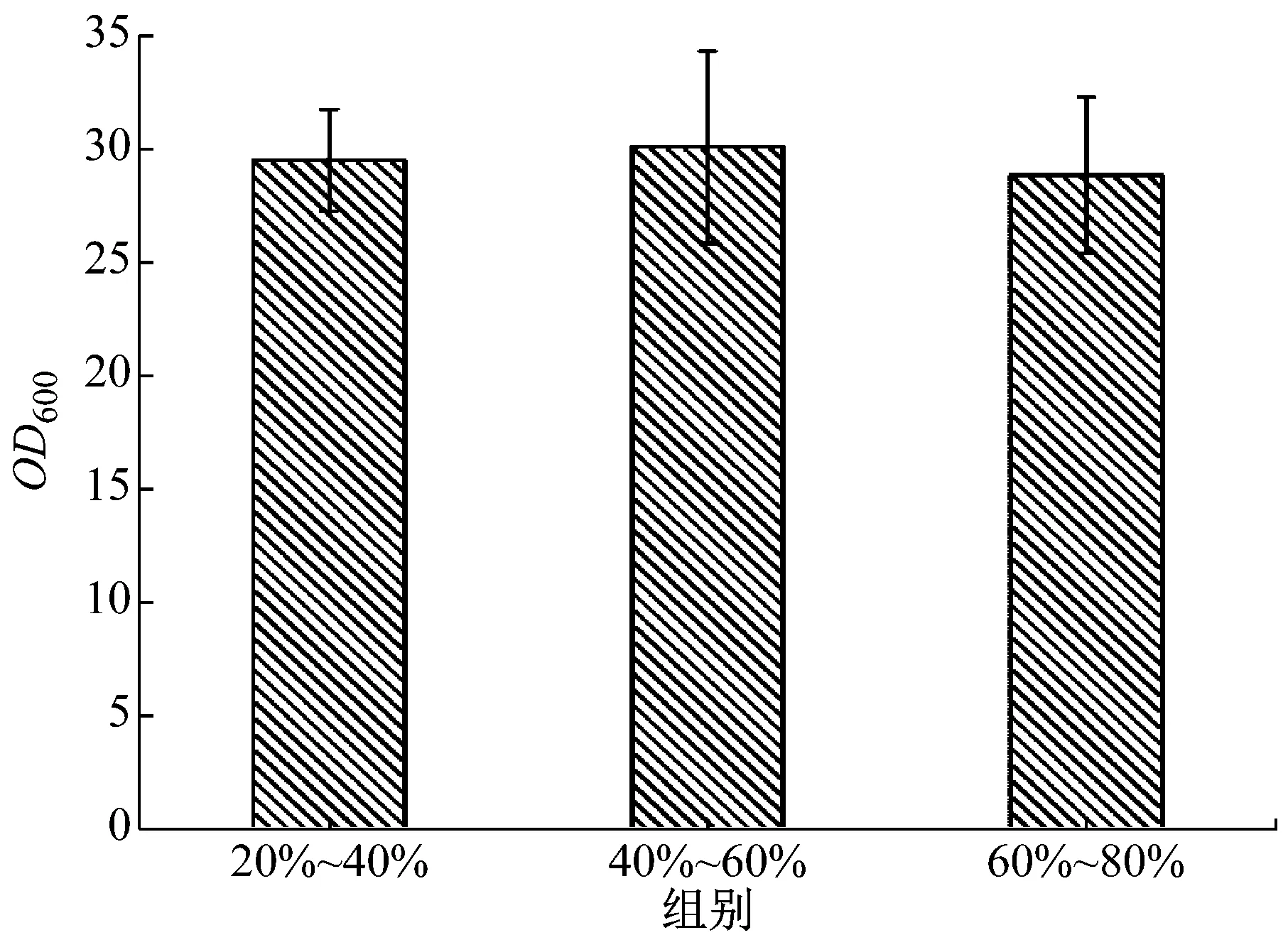
圖4 20%~40%、40%~60%和60%~80%組別菌的濃度
2.3 畢赤酵母PAOX2上游調控序列分析
測序結果顯示共有兩株菌株(20-16和20-20)的PAOX2發生了突變,其與NCBI公布(模板質粒的PAOX2已測序,顯示其與NCBI公布的PAOX2完全一致)的PAOX2序列比對結果(如圖5所示)。PAOX2共有1 550個堿基,突變型菌株20-16在702(即-849)位置發生堿基T的插入導致其表達木聚糖酶活力為對照組菌株的36%突變型菌株20-20在789(即-762)位置插入了堿基A,其表達木聚糖酶的酶活為對照組菌株的10%。OHI等[15]鑒定出一個上游激活元件(UAS)和兩個上游抑制元件(URS)。UAS位于PAOX2的-341至-273之間,其中,處于-337至-313位置的序列GATAGGCTATTTTTGTCGCATAAAT是PAOX2UAS的重要組成部分,且處于PAOX2的-293至-283之間的序列不參與轉錄調控,PAOX2不會因為此片段的缺失而提高表達能力。兩個URS分別位于PAOX2的-780至-342之間和-255至-215之間。本研究的結果表明在-849~-762之間的堿基序列也對PAOX2的轉錄有重要作用。

圖5 畢赤酵母PAOX2與測序菌株的PAOX2的比對結果
本研究中仍有大量未發生突變的PAOX2的片段導致木聚糖酶活力顯著下降,這可能由于大批量培養重組畢赤酵母可能會導致平行性出現較大差異,重組畢赤酵母表達木聚糖酶活力產生波動。
3 結論
通過易錯PCR的方式對PAOX2上游調控序列進行隨機突變,構建了PAOX2突變文庫,篩選了850株重組菌。測序結果表明突變型菌株在-847插入堿基A或者-788插入堿基T分別使木聚糖酶活力下降為10%和36%。這說明-847 ~-788片段對PAOX2的轉錄有重要作用。
