快速制備不銹鋼自支撐析氧催化劑的性能研究
2021-07-03 07:04:24李玉平熊圖治韋晶晶BalogunMuhammadSadeeq
湖南大學學報(自然科學版) 2021年6期
關鍵詞:催化劑
李玉平,熊圖治,韋晶晶,Balogun Muhammad-Sadeeq
(湖南大學材料科學與工程學院,湖南長沙 410082)
水分解技術可以分為析氫反應(HER)和析氧反應(OER)兩個電化學半反應.使用催化劑降低反應能壘,可使水分解制氫更符合實際需求.現常用的OER 和HER 催化劑有Ir 基、Ru 基和Pt 基等貴金屬化合物催化劑[1-3],它們雖然能較大幅度降低水分解所需的電壓,但因資源稀缺、成本高昂以及穩定性較差而影響了其規模化使用[4-5].正因為如此,過渡金屬電催化劑已備受人們青睞.有些過渡金屬催化劑的催化性能已經接近甚至優于貴金屬催化劑[6-8].
不銹鋼網因其具有良好的導電性、在堿性介質正電位下的高穩定性、自身的三維結構、適當的柔性及低廉的價格而成為電化學過程(如OER)的理想基體之一[9-10],通過適當處理后,可直接用做OER 催化劑.例如Balogun 等[11]曾用LiCl·H2O 和H3PO4在120 ℃腐蝕AISI-304 不銹鋼網并將其在N2氣氛下退火后直接用于OER 催化.在10 mA·cm-2處的過電位為278 mV,塔菲爾斜率為83 mV·dec-1,電解1×106s 后,催化性能沒有發生明顯變化.但因需要腐蝕和退火處理,其制備周期至少需要4 h 以上,不利于規模化快速生產,并且OER 性能仍然有待提升.Zou等人[12]將負載有導電Ni3S2納米片的泡沫鎳(Ni3S2/NF)作為前驅體,將其置于含Fe3+的預熱溶液中5 s后,便可以得到生長有非晶態Ni-Fe 雙金屬氫氧化物的Ni-Fe-OH@Ni3S2/NF,在過電位為167 mV 時具有10 mA·cm-2的電流密度,過電位為300 mV 時,電流密度可以達到100 mA·cm-2.盡管Ni-Fe 雙金屬氫氧化物的制備特別迅速,但是Ni3S2/NF 前驅體仍然需要在反應釜反應5 h 以上才能得到.
本文主要研制了一種便于規模化快速生產且具有較高OER 催化性能的自支撐電催化劑(SS/Ni-OH2M-120s).將AISI-304 不銹鋼網浸入含有2 mol·L-1Ni2+的沸騰溶液中120 s,便可以得到在10 mA·cm-2處過電位僅為214 mV 的OER 自支撐電催化劑,比不銹鋼網降低接近127 mV(不銹鋼網在10 mA·cm-2處的過電位為341 mV),并且其催化性能在20 mA·cm-2的電流密度下恒電位極化10 h 沒有發生明顯變化.將之與Pt 網電極組成全分解水裝置后,10 mA·cm-2處電位僅為1.61 V.同時,通過更改溶液離子成分和降低溶液離子濃度等方式,發現將不銹鋼網浸入含有Fe3+或者Ni2+和Fe3+的溶液中,也能使其催化性能得到極大的提高,從而驗證了該方法的普遍適應性.
1 實 驗
1.1 實驗藥品
氯化鎳(NiCl2·6H2O,99%,分析純,上海麥克林生化科技有限公司);三氯化鐵(FeCl3·6H2O,99%,分析純,上海麥克林生化科技有限公司);硝酸鈉(NaNO3,99%,分析純,國藥集團化學試劑有限公司);AISI-304 不銹鋼網(孔徑13 μm,佛山市廣美不銹鋼有限公司).
1.2 實驗流程
304 不銹鋼網(SS)經丙酮、乙醇和超純水超聲清洗數次后,在60 ℃烘箱中干燥備用.按實驗配比(表1)將氯化鎳溶于已溶解硝酸鈉(2.49 mmol/L)的沸騰溶液中,溶液再次沸騰后將已清潔的不銹鋼網(1.5 cm×2 cm)浸入溶液,分別保持10 s、60 s、120 s 后取出,于60 ℃烘箱干燥便可以得到OER 自支撐電催化劑.
1.3 材料表征
用場發射掃描電子顯微鏡(FESEM,Quanta 400/INCA/HKL)觀察樣品形貌.用X 射線衍射儀(XRD;Shimadzu X 射線衍射儀6000,Cu Kα,Tokyo,Japan)以8(°)·min-1的掃描速率測定樣品的物相.用辰華760E 電化學工作站測定所有電化學性能,將制備的樣品直接用作自支撐工作電極,Pt 絲作為對電極,Ag/AgCl(飽和溶液)作為參比電極,1 mol·L-1的KOH溶液為電解液,掃描速率為2 mV·s-1.為精確確定催化劑的催化區域,用于電化學測試的樣品均使用環氧樹脂進行處理,留下1 cm2的正方形區域用于電化學催化,在另一端留下小部分區域用于歐姆接觸.
2 結果與討論
以具有良好韌性的奧氏體相為主的304 型不銹鋼網(Cr0.19Fe0.7Ni0.11)作為催化劑基體,將其浸入含Ni2+的沸騰溶液進行表面處理,期望能在表面快速生成Ni 金屬氫氧化物或者Ni/Fe 雙金屬氫氧化物,得到高性能OER 自支撐電催化劑.
圖1 為樣品的物相組成及表面形貌.由圖1(a)可 知,不銹鋼網、SS/Ni-OH0.5M-120s 和SS/Ni-OH2M-120s 都有與奧氏體相相對應的峰(也稱為γ相)(PDF 卡#33-0397).同時SS/Ni-OH0.5M-120s和SS/Ni-OH2M-120s 在21°左右出現了新的峰,該新峰可能是鎳的氫氧化物(Ni-OH)[13-15],說明不銹鋼網經短時間處理后,形成了鎳金屬氫氧化物(Ni-OH).
由圖1(b)(d)空白不銹鋼網的掃描電鏡照片可知,不銹鋼網表面光滑平整,無其他的雜質附著.圖1(c)(e)為SS/Ni-OH2M-120s 的電子顯微鏡照片,放大500 倍后,樣品表面依舊相當規整,但是邊緣存在著凹凸不平的現象;放大至50 000 倍后可以明顯看出,經處理的不銹鋼表面存在均勻分布的顆粒,結合XRD 結果可知,這些顆粒應該是Ni-OH,并且顆粒間通過松散交聯形成了三維微孔結構.Ni-OH 本身優異的OER 活性[16-17]、三維微孔結構有利于氣體脫離以及增加比表面積等[18-20]性質,預計將大幅度提高OER 催化性能.
圖2(a)為不銹鋼網經含2 mol·L-1Ni2+沸騰溶液處理120 s(SS/Ni-OH2M-120s)后的LSV 曲線和不銹鋼網自身的LSV 曲線.由圖可以看出,SS/Ni-OH2M-120s 的催化性能明顯優于不銹鋼網,僅需要214 mV 的過電位便可以提供10 mA·cm-2的電流密度,而不銹鋼網則需要高達341 mV 的過電位才能得到同樣的電流密度,比SS/Ni-OH2M-120s高127 mV.
圖2(b)為不銹鋼網經不同Ni2+濃度溶液處理120 s 后的LSV 曲線.由圖可知,在這些濃度下,不同樣品的OER 性能和LSV 曲線斜率均基本接近,但是SS/Ni-OH2M-120s 略好于SS/Ni-OH0.5M-120s 和SS/Ni-OH1M-120s.這3 種濃度下的LSV 曲線都在150~160 mV 存在一個陽極氧化峰,而且SS/Ni-OH2M-120s 的陽極氧化峰明顯高于SS/Ni-OH0.5M-120s 和SS/Ni-OH1M-120s,因此這個峰可能與鎳的陽極峰有關.
圖2(c)為不銹鋼網分別在0.37 mmol·L-1Ni2+(SS/Ni-OH-120s)、0.37 mmol·L-1Fe3+(SS/Fe-OH-120s)、0.37 mmol·L-1Fe3+和0.37 mmol·L-1Ni2+(SS/Fe-Ni-OH-120s)溶液中處理120 s 后的LSV 曲線.由圖可以看出,在10 mA·cm-2電流密度下的過電位仍然可以比不銹鋼網降低至少80 mV.并且SS/Ni-OH-120s 和SS/Fe-Ni-OH-120s 的OER 性能優于SS/Fe-OH-120s,可能原因是,鎳的氫氧化物和鎳鐵雙金屬氫氧化都能大幅度提升OER 催化劑的催化性能,但是當Fe 單獨存在時,較多的鐵會降低催化劑催化性能[21],因此在3 組樣品中,SS/Fe-OH-120s的催化性能最差,但是由于其能形成Fe 金屬氫氧化物,且不銹鋼本身就存在鎳元素,也有可能形成微量鎳鐵雙金屬氫氧化物,因此性能(10 mA·cm-2電流密度下的過電位為293 mV)仍然明顯優于不銹鋼網.
為探究SS/Ni-OH2M-120s 經溶液處理120 s后,性能比空白不銹鋼明顯提升的機理,對SS/Ni-OH2M-120s 和空白不銹鋼進行了電化學活性表面積和塔菲爾斜率的表征.
圖3(a)和圖3(b)分別為SS/Ni-OH2M-120s 和空白不銹鋼網試樣在不同掃描速率下的CV 曲線,圖3(c)為二者的雙層電容.由于電化學活性表面積與雙層電容成正比,由圖可知,SS/Ni-OH2M-120s 的雙層電容為0.161 mF·cm-2,比空白試樣的0.12 mF·cm-2高34%.因此其具有更大的電化學活性表面積.
圖3(d)為SS/Ni-OH2M-120s 和空白不銹鋼試樣的塔菲爾斜率.通過塔菲爾斜率分析可以更好地理解催化劑的催化動力學.OER 反應過程通常由4個反應步驟組成,速率決定步驟取決于反應步驟中的電子轉移效率,據文獻報道[4],如果傳遞系數為0.5,相應的塔菲爾斜率為120 mV·dec-1.速率決定步驟為:
M+OH-→MOH
若傳遞系數為1.0,則相應的塔菲爾斜率為60 mV·dec-1,速率決定步驟為:
MOH+OH-→MO+H2O
塔菲爾斜率由塔菲爾方程得出:
η=b lg j+a
其中b 和j 分別為塔菲爾斜率和電流密度.由圖3(d)可知,SS/Ni-OH2M-120s 的塔菲爾斜率為34.84 mV·dec-1,不銹鋼網的塔菲爾斜率為46.06 mV·dec-1,兩者都比較小且相對接近60 mV·dec-1,所以速率決定步驟可能是:
MOH+OH-→MO+H2O
更大的電化學活性表面積和更優異的電化學動力學導致SS/Ni-OH2M-120s 的電催化析氧性能相比空白不銹鋼有了巨大的提升.
圖4 為催化劑催化性能的穩定性測試.由圖4(a)(b)可知,恒電位極化10 h 和恒電流極化30 h后,SS/Ni-OH2M-120s 試樣的電流密度沒有出現明顯變化.圖4(c)為穩定性測試前后的LSV 曲線及穩定性測試后的微觀形貌,由圖可知,催化劑在20 mA·cm-2的電流密度下持續工作10 h 后,10 mA·cm-2處的過電位為218 mV,與穩定性測試前相比,過電位沒有發生明顯變化,顯示了SS/Ni-OH2M-120s優良的穩定性.通過與圖1(e)對比可以看出,穩定性測試后樣品表面堆積的鎳氫氧化物以及顆粒間的微孔結構并未發生明顯變化,仍然保持穩定性測試之前的基本形貌,表明了該催化劑優異的形貌穩定性.同時,通過圖4(d)穩定性測試前后XRD 圖譜的對比,可以發現,穩定性測試前后沒有發生物相變化,但是在穩定性測試后,樣品在21°左右的衍射峰有所增強,金屬基催化劑在OER 過程中,可能因為陽極氧化作用,在其表面會形成一定量的金屬氫氧化物[22].因此21°左右峰值的變強,可能是SS/Ni-OH2M-120s樣品在OER 過程中,其表面新形成了金屬氫氧化物,并且此峰值的變化,從另一方面驗證了21°的峰是鎳金屬氫氧化物.
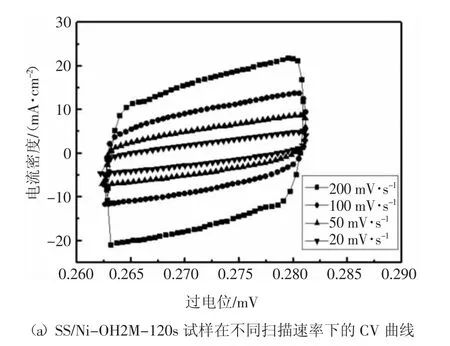
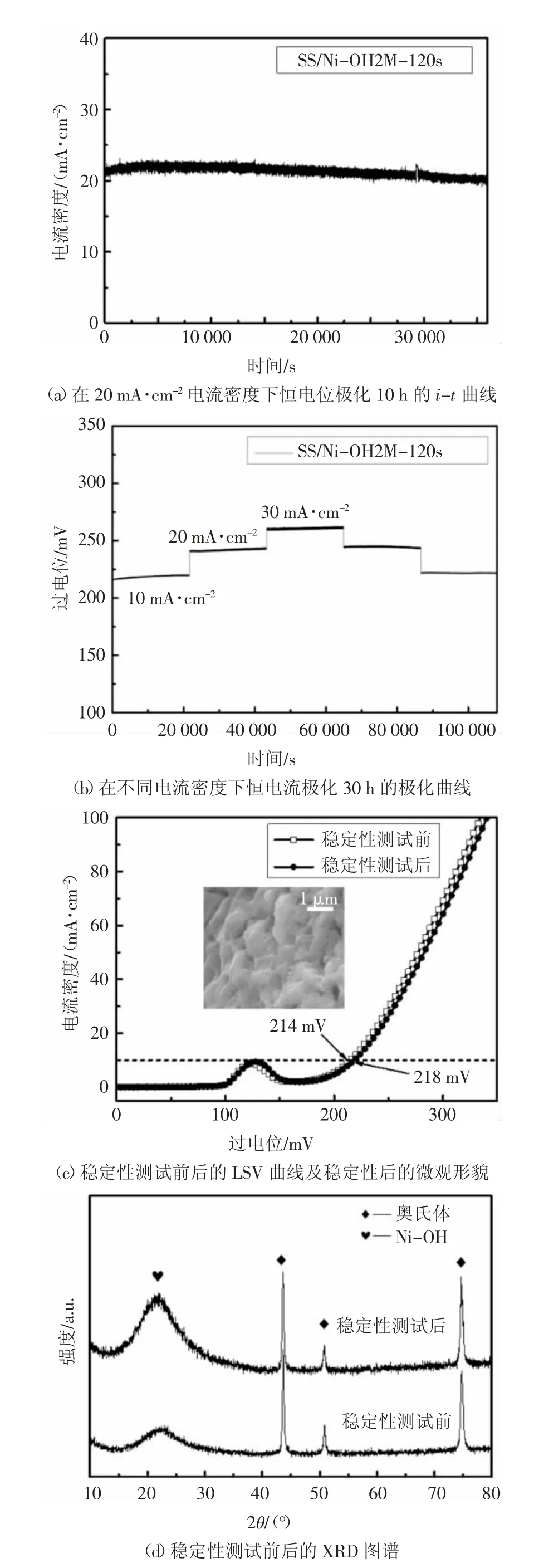
圖4 穩定性測試Fig.4 Stability test
圖5 為SS/Ni-OH2M-120s 試樣電催化劑的擴展性制備演示及其全分解水的LSV 曲線.圖5(a)(b)是一次性處理304 不銹鋼網后的實際操作圖,制備試樣的大小擴大為10 cm×10 cm,將其浸入含2 mol·L-1Ni2+的沸騰溶液處理120 s 后,置于烘箱60 ℃烘干,得到一次性制備的自支撐OER 催化劑.從新制得的試樣上取出1 cm×0.5 cm 的小片,用環氧樹脂密封,與Pt 網電極組裝成全分解水裝置(表示為Pt-Mesh//SS/Ni-OH2M-120s),并將其性能與商用IrO2和Pt 網陰極全分解水器件進行比較(表示為Pt-Mesh//IrO2/SS).圖5(c)(d)分別為全分解水裝置實際水電解圖片和Pt-Mesh//SS/Ni-OH2M-120s 與Pt-Mesh//IrO2/SS 的LSV 曲線.由圖5(d)可知,Pt-Mesh//SS/Ni-OH2M-120s 分解水的能力明顯優于Pt-Mesh//IrO2/SS,在10 mA·cm-2的電流密度下,其電壓為1.61 V,而Pt-Mesh//IrO2/SS 的電壓為1.84 V,相比降低了0.23 V.通過對SS/Ni-OH2M-120s 進行擴大化制備,并將其用作全分解水裝置后,可以發現,此方法適合規模化生產.

3 結論
1)將AISI-304 不銹鋼網在含有2 mol·L-1Ni2+的沸騰溶液中經過極短時間的浸泡處理后,其表面會被彼此堆積形成三維微納結構的鎳金屬氫氧化物覆蓋.得到在10 mA·cm-2電流密度時,過電位僅為214 mV,20 mA·cm-2電流密度下恒電位極化10 h,以及不同電流密度下恒電位極化30 h 后,催化性能和物相組成等基本不變的自支撐催化劑(SS/Ni-OH2M-120s).
2)將SS/Ni-OH2M-120s 和Pt 網電極組裝成的全分解水裝置在10 mA·cm-2電流密度下的電壓為1.61 V,比Pt-Mesh//IrO2/SS 電壓降低了0.23 V.
3)通過降低溶液離子含量、調整溶液離子組分和擴大制備面積制備等試驗,驗證了此高性能OER自支撐催化劑可用于規模化快速制備.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
