能量合成障礙在阿爾茨海默病發病中的作用
2021-08-10 00:33:56牛芬溪
中國藥理學通報 2021年8期
牛芬溪,謝 碩,劉 悅,方 芳
(北京中醫藥大學中藥學院,北京 102488)
AD是一種神經退行性疾病,也是目前最為常見的老年癡呆病。據2018年報道顯示,全球目前有5 000萬人罹患AD,到2030年預計增加到8 200萬人,而2050年患者總數將增加到現在的3倍,高達1.52億人。2018年全球在治療AD上的花費估計在1萬億美元,2030年總花費預計將達到2萬億美元,給患者的家庭和社會造成了極大的精神和經濟負擔[1]。AD患者的臨床表現以漸行性記憶力減退,認知功能損害為主,但是其早期癥狀并不明顯,發病隱密,導致病因學研究進展緩慢。AD的病理學特征主要包括細胞內高度磷酸化的tau蛋白組成的神經纖維纏結(neurofibrillary tangles,NFTs)、細胞外Aβ錯誤折疊形成的老年斑塊(senile plaques,SPs)、神經元和突觸的丟失。雖然Aβ和過度磷酸化的tau蛋白被認為是導致神經元毒性的重要原因,但針對二者研發的抗AD藥物的臨床試驗效果并不理想。目前尚未發現能夠根治AD的藥物,越來越多的研究者開始關注AD早期的病理變化,治療策略也轉向多樣化。
能量代謝是生物體維持細胞穩態,保證正常生命活動的物質基礎。腦組織高代謝速率和神經元維持靜息電位和進行突觸傳遞等特點均提示大腦是一個極度需要能量的器官。研究表明,至少7%的早發型AD、20%晚發型AD及35%-50%晚期AD患者的大腦中ATP生成減少,特別是神經元的能量合成減少早于AD認知功能和病理特征的出現,且伴隨疾病進展,能量合成逐漸減少并能促進Aβ沉積等病理變化的形成[2],故能量合成障礙被認為是AD早期特征性病理改變之一。由于血腦屏障的限制,葡萄糖是哺乳動物大腦新陳代謝的主要能量來源,同時葡萄糖代謝還參與神經活性物質(如谷氨酸、天冬氨酸、甘氨酸等)生成和神經細胞的增殖,對維持大腦突觸重構、增強認知功能非常重要[3]。本文圍繞腦血流量和葡萄糖的攝取、利用及代謝調控等環節,綜述能量合成障礙在AD中的研究進展。
1 腦血流量降低
腦血流量與大腦葡萄糖和氧的供給有關。非臨床期和早期的AD腦組織低灌注比代謝減慢更明顯。研究發現,某些腦區腦血流量減少50%時可以降低中樞神經系統中Na+-K+ATPase活性,降低ATP合成,影響蛋白穩態。AD早期血流量降低早于突觸和神經元丟失,提示慢性血流降低可能是AD認知功能障礙的重要原因。AD患者腦微血管結構損傷研究發現,AD病人早期腦內毛細血管,特別是在淀粉樣斑塊周圍的毛細血管出現收縮現象,與腦血流降低有關。腦組織中毛細血管收縮除受血管平滑肌調控外,還與周細胞有關。通過測定毛細血管的傳遞時間及其異質性,可以看出AD患者毛細血管周細胞介導的血流控制被中斷。血流量減少導致腦組織缺血缺氧,除直接減少腦組織葡萄糖供給外,還會引起氧化應激和活化內皮素受體,加速Aβ沉積,引起神經元和BBB損傷[4]。Aβ本身也可以直接誘導周細胞介導的毛細血管收縮,減少腦組織血流量,干擾能量合成,促進AD進展。因此,在神經元、突觸等未明顯損傷前,干擾周細胞介導的毛細血管收縮和毛細血管堵塞,增加腦組織血流量,改善腦能量合成障礙,恢復認知功能已成為新的預防AD的策略。
2 葡萄糖攝取障礙
腦神經元并不能直接合成或儲存葡萄糖,它們完全依賴于從外部持續攝取葡萄糖以滿足自身功能。由于葡萄糖是一種極性的親水分子,不能隨意透過血腦屏障,它的轉運在很大程度上是依靠由SLC5基因編碼的鈉依賴性葡萄糖轉運體(sodium depedent glucose transporter,SGLTs)和由SLC2基因編碼的鈉非依賴性葡萄糖轉運體(glucose transporter,GLUTs)來實現的。到目前為止,在人體組織中已發現14個鈉非依賴性葡萄糖轉運蛋白和12個鈉依賴性葡萄糖轉運蛋白,人腦表達10個GLUT蛋白和10個SGLTs蛋白[5]。研究發現,GLUT1主要負責將葡萄糖從血液運輸到大腦的細胞外空間,包括通過55ku GLUT1的跨內皮細胞的轉運和45 ku GLUT1向圍繞在突觸部位的星形膠質細胞的轉運,而 GLUT3則主要負責將葡萄糖從細胞外空間運輸到神經元,且轉運效率遠高于GLUT1。但是大腦葡萄糖是首先通過GLUT1進入星形膠質細胞,還是通過GLUT3直接從血管輸送到神經元,目前還沒有定論。也有研究發現,45 ku GLUT1在少突膠質細胞表達,而非小膠質細胞。除此之外,GLUT2可能主要在星形膠質細胞中表達,也存在于下丘腦神經元中,發揮大腦葡萄糖傳感器的作用。GLUT4是一種胰島素敏感轉運蛋白,腦組織高能量需求時,GLUT4提供快速葡萄糖供給[6]。有研究表明,腦葡萄糖攝取量下降和胰島素敏感轉運蛋白表達下降之間具有很強的相關性。GLUT4的活性上調將導致組織對葡萄糖攝取利用的增加。
臨床前和臨床研究均顯示,低水平的葡萄糖攝取與年齡相關的認知障礙有關,更容易出現MCI或AD。腦組織中葡萄糖攝取減少伴隨著腦組織結構和功能變化。正電子發射斷層掃描(PET)研究發現,AD患者某些腦區,如海馬、扣帶回、顳葉、頂葉和額葉均顯示明顯的葡萄糖攝取減少,GLUT1和GLUT3蛋白水平明顯降低,且與Tau蛋白高度磷酸化水平相關[7]。18月齡APP/PS1小鼠海馬區GLUT1表達明顯比同月齡野生型小鼠減少[8]。在雄性3xTg-AD小鼠中,神經元GLUT3和GLUT4表達下降;在雌性3xTg-AD小鼠中,大腦葡萄糖攝取與GLUT3表達降低一致,但與GLUT4的表達無關[9]。在兩種淀粉樣變性小鼠模型(Tg2576和APP/PS1)的額葉皮質中,老齡小鼠和SAMP8小鼠海馬區中均發現GLUT1和GLUT3的表達減少[10]。
已有許多研究表明,通過調控GLUT可以改善能量代謝,進而改善AD病理。Mana等[11]發現,中藥復方GAPT(由人參、石菖蒲、多根、姜黃等草本提取物混合而成)通過增加腦葡萄糖攝取、增加葡萄糖轉運和改善胰島素信號通路改善了APP/PS1轉基因小鼠的腦葡萄糖代謝損傷。Chen等[12]觀察到人參皂苷的腸道菌群代謝產物Compound K通過促進GLUT的活性增加了被Aβ誘導損傷的HT22細胞的ATP生成量。Pang等[13]發現規律運動使得AD模型小鼠腦內GLUT1和GLUT3表達水平提高,ATP生成量增加,突觸數目增多,線粒體嵴和邊緣清晰完整,提示規律運動后GLUT1和GLUT3在蛋白水平的表達變化是AD模型小鼠能量代謝適應的重要組成部分。
3 葡萄糖代謝障礙
進入神經細胞中的葡萄糖主要的代謝途徑包括糖酵解和有氧氧化。糖酵解途徑將葡萄糖轉變為丙酮酸進入線粒體,在三羧酸循環(tricarboxylic acid cycle,TCA cycle)中進一步代謝,通過氧化磷酸化產生三磷酸腺苷(adenosine triphosphate,ATP)。有研究發現,健康老年人中認知能力與葡萄糖利用有密切關系。應用18F-FDG PET技術可以檢測到AD早期伴隨著大腦對葡萄糖利用的降低和能量缺失,在無癥狀家族型AD的高危人群中也檢測到葡萄糖代謝顯著降低。甚至有報道發現AD患者在出現認知功能減退和腦組織結構無明顯改變前,大腦皮層的糖代謝已經出現異常變化[14]。
3.1 葡萄糖酵解障礙進入細胞內的葡萄糖經己糖激酶、6-磷酸果糖激酶、丙酮酸激酶等代謝成丙酮酸,此過程中每消耗1分子葡萄糖產生2分子ATP,雖然產生的能量較低,但該過程產生能量的速度快,特別是在突觸部位葡萄糖酵解被認為是重要的能量來源,參與突觸可塑性。特別是在AD腦中能量需求增加,伴有缺氧和氧化應激,神經細胞更多的依賴糖酵解來合成ATP,且腦組織葡萄糖酵解與腦內Aβ沉積呈顯著的相關性。AD患者腦內和微血管中己糖激酶活性降低,同樣在SAMP8小鼠皮層、海馬和紋狀體中也觀察到葡萄糖代謝障礙與己糖激酶活性降低有關。此外,AD患者腦能量減少也與葡萄糖酵解過程中的磷酸丙糖異構酶、磷酸甘油酸變位酶活性異常或表達降低有關[15]。
3.2 線粒體功能障礙線粒體是一種存在于大多數細胞中的雙層膜細胞器,負責從葡萄糖代謝、脂肪酸氧化和呼吸等生化過程中產生大量的細胞能量,并以ATP的形式表現出來。葡萄糖酵解產物丙酮酸進入線粒體后經丙酮酸脫氫酶催化產生乙酰輔酶A,除參與能量合成外,還是合成與學習記憶密切相關的神經遞質乙酰膽堿的原料,也可通過天冬氨酸-草酰乙酸合成N-乙酰天冬氨酸參與神經髓鞘磷脂的生物合成。α-酮戊二酸脫氫酶復合物可以轉變成谷氨酸和GABA,參與維持突觸可塑性。大量的研究結果顯示,線粒體損傷是AD的主要病理特征,在家族型和散在型AD病人和多種AD動物模型中均可見不正常的線粒體、TCA循環和OXPHOS受損、表現為線粒體數量和體積減小,線粒體膜電位、線粒體呼吸和葡萄糖代謝降低,相關代謝酶如丙酮酸脫氫酶復合物(pyruvate dehydrogenase complex,PDHC)、α-酮戊二酸脫氫酶復合物(α-ketoglutarate dehydrogenase complex,KDHC)、琥珀酸脫氫酶和細胞色素c氧化酶(cytochrome c oxidase,COX)、異檸檬酸脫氫酶、琥珀酸脫氫酶等的表達和活性降低[16]。用13C標記葡萄糖,利用核磁共振波譜分析發現,APPswe-PS1dE9和3xTG-AD小鼠腦內葡糖糖利用明顯降低,TCA循環代謝產物谷氨酸、GABA和谷氨酰胺減少,提示這些小鼠大腦中葡萄糖氧化和神經遞質循環受損[17]。研究發現APP可以在線粒體中經細胞質中β-分泌酶(β-site APP cleaving enzyme,BACE1)和線粒體上的γ-secretase的剪切產生Aβ。Aβ可以與線粒體膜上蛋白結合進入線粒體,與Aβ結合乙醇脫氫酶(Aβ-binding alcohol dehydrogenase,ABAD)結合導致線粒體功能障礙,降低COX活性并損傷線粒體ETC,從而導致活性氧生成增加和ATP生成減少,生成的活性氧進一步加重線粒體功能障礙,如此形成一個惡性循環[18]。轉染AD患者mtDNA的細胞內活性氧明顯增加,AD患者腦線粒體mtDNA含被氧化的堿基含量高于核DNA,提示出現線粒體功能障礙和氧化損傷。“線粒體級聯學說”認為線粒體功能障礙是能量合成障礙的重要環節,也是AD發病早期的最主要特征。促進線粒體合成,恢復線粒體功能,改善大腦能量代謝被認為是一種抗AD的治療策略[19]。見Fig 1。
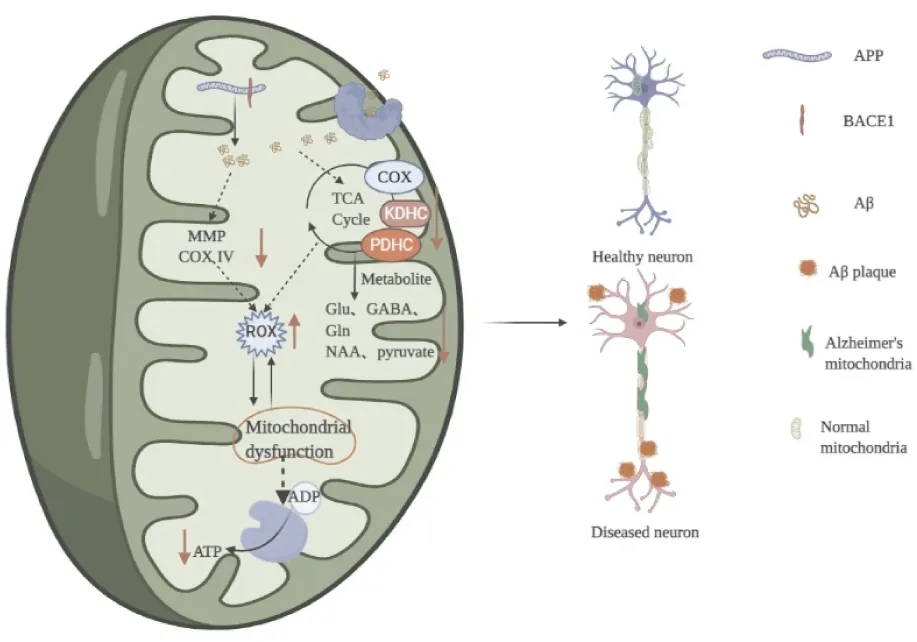
Fig 1 Induced mitochondria by Aβ led to impaired glucose metabolism, decreased energy synthesis and impaired neuronal structure and function
4 葡萄糖能量合成相關的調控分子缺陷
4.1 胰島素/胰島素樣生長因子1信號 (insulin/insulin-like growth factor-1 signaling,IIS) 缺陷胰島素作為一種重要的生長因子,不僅能通過血腦屏障,也能在大腦內合成,參與調節細胞生長、能量利用、線粒體功能、自噬、氧化應激、突觸可塑性和認知功能。胰島素樣生長因子1(insulin-like growth factor-1,IGF-1)又稱促生長因子,與胰島素分子結構類似,可增加對胰島素的靈敏度,促進葡萄糖的利用。胰島素和IGF-1與兩個高度相關的酪氨酸激酶受體-胰島素受體(insulin receptor,IR)和IGF-1受體(IGF-1R)結合后,磷酸化IR受體和與其共定位的胰島素受體底物(insulin receptor substrate,IRS),活化的IRS通過激活PI3K/Akt信號通路參與葡萄糖攝取、神經元存活和突觸可塑性調節[20]。另外,胰島素和IR與GLUT4共同定位于海馬和脈絡叢神經元,提示GLUT4在胰島素/IR誘導的葡萄糖攝取中具有重要作用。Pharaoh等[21]研究發現IGF-1缺乏會損害雄性Igf1f/f小鼠依賴海馬的空間獲得和反轉學習,海馬線粒體氧化磷酸化偶聯效率和皮層ATP水平降低。Logan等[22]利用原代培養的星形膠質細胞,通過減少IGF受體表達來降低IGF-1信號,結果發現ATP的合成顯著受損。IGFR缺陷的星形膠質細胞還表現出葡萄糖攝取受損、線粒體結構和功能改變。故IIS信號通路在調節葡萄糖代謝,維持能量穩態等方面發揮著不可替代的作用,見Fig 2。
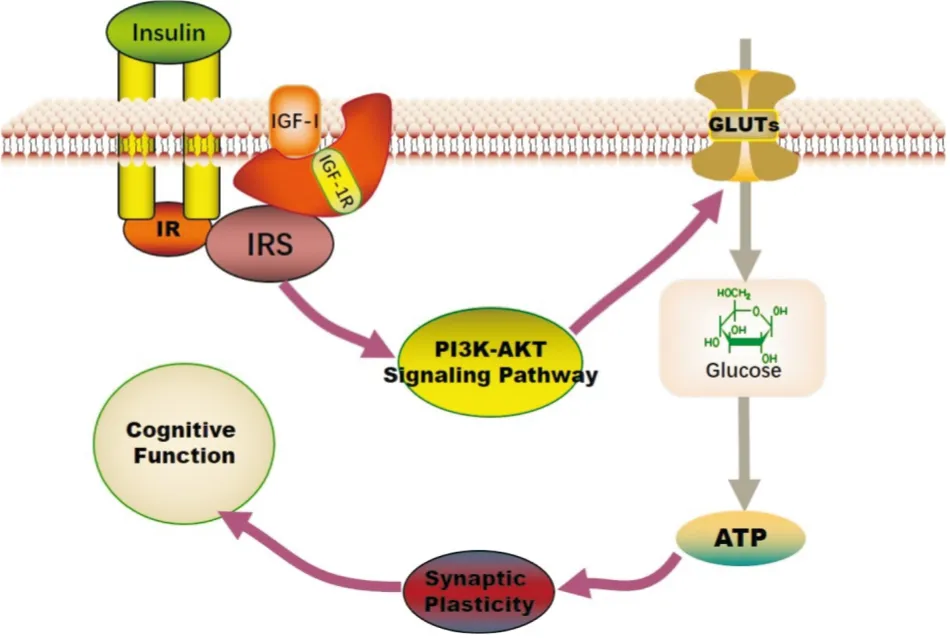
Fig 2 Regulation of insulin signaling pathways on bioenergetic synthesis of glucose
在AD患者腦中和Aβ 1-42寡聚體誘導的人星形膠質細胞模型中均發現胰島素、IGF-1及其受體和下游信號分子(PI3K/Akt)的mRNA和蛋白質出現了顯著地下降。有研究發現,3xTg-AD小鼠早在1個月時就表現出糖耐量受損,約14個月時檢測到斑塊、18個月時出現過度磷酸化tau聚集和認知功能下降,且出現AD病理癥狀前就伴隨著血漿胰島素水平的下降,而PI3K/AKT信號通路在18-20月齡的3xTg-AD小鼠中發生了改變[23]。6月齡APP/PS1小鼠表現出認知能力受損,葡萄糖耐受不良,海馬中胰島素受體轉錄降低,線粒體功能障礙,能量代謝異常均早于淀粉樣斑塊形成。腦室注射鏈尿佐菌素的大鼠出現胰島素抵抗,腦葡萄糖代謝及能量代謝異常,進行性學習記憶能力降低。Aβ可以與胰島素競爭與受體結合引起胰島素抵抗,導致葡萄糖代謝紊亂,胰島素信號通路受損可抑制胰島素降解酶的表達和活性,增加Aβ沉積,導致AD發生[24]。這些研究提示IIS信號通路的障礙,包括腦胰島素抵抗、IGF-1抵抗和IRS1功能障礙會影響葡萄糖代謝,引起能量合成障礙。Lee等研究發現來自闊葉山麥冬的新化合物(LP9M80-H)正是通過誘導IRS2表達增加,通過PI3K/Akt促進神經元葡萄糖轉運體的生物合成,增加葡萄糖的攝取和利用,導致ATP的產生增加[25]。胰高血糖素樣肽(GLP-1)是腦內神經元產生參與調節血糖水平的物質。GLP-1以依賴血漿葡萄糖水平的方式改變血-腦葡萄糖轉運和腦內葡萄糖代謝率,具有改善胰島素抵抗的作用。研究發現GLP-1能調節星形膠質細胞和5xFAD小鼠皮層的糖酵解作用,減少氧化損傷,增加ATP產生,改善小鼠認知功能[26]。
4.2 AMPK信號通路缺陷AMPK(adenosine 5′-monophosphate (AMP)-activated protein kinase,AMPK)是一種在真核細胞中保守的細胞能量傳感器,它能抑制消耗能量的合成代謝過程,促進產生能量的分解代謝過程,任何通過干擾ATP合成來擾亂能量平衡的代謝壓力都會激活AMPK。當線粒體功能發生障礙時,細胞中AMP/ATP比例將會上調,導致AMPK被激活。活化的AMPK可以觸發GLUT的膜移位,增加葡萄糖攝取,促進葡萄糖酵解和ATP產生。特別是AMPK可磷酸化并直接激活過氧化物酶體增殖物激活受體γ輔激活因子1α(peroxisome proliferator-activated receptor γ coactivator-1α,PGC-1α)。PGC-1α是線粒體生成的主要調控因子,也是能量代謝的關鍵調節因子。該信號通路除了能夠發揮調節大腦能量代謝,改善大腦的攝取速率和代謝率,防止神經元退變和死亡的作用之外,還可調節BACE1的轉錄、合成和活性,減少APP代謝生成Aβ,進而減少淀粉樣斑塊的形成,防止AD的發生發展[27]。臨床研究表明,AD患者AMPK活性降低,PGC-1α的mRNA及蛋白水平均低于健康人群。白藜蘆醇通過增強AMPK和PGC1α活性,激活SIRT1促進線粒體發生,能減輕Aβ1-42誘導大鼠學習記憶障礙和APP/PS1的小鼠腦內淀粉樣斑塊的產生和氧化應激反應[28]。
4.3 缺氧誘導因子表達降低缺氧誘導因子1(hypoxia inducible factor-1,HIF-1)是GLUT1和GLUT3的主要調節因子,在正常糖代謝中起著重要的作用[29]。在AD患者腦內可以觀察到HIF-1下調,可能是由于氧化應激增加破壞了HIF-1的穩定性,因此,GLUT1和GLUT3水平的降低可能是由于HIF-1下調所致。研究發現通過CRISPR/Cas9技術對HIF-1α和GLUT1雙基因敲除抑制了PI3K/Akt/mTOR通路,導致葡萄糖攝取減少[29]。
4.4 雌激素缺乏雌激素是一種類固醇激素,參與葡萄糖轉運、糖酵解和有氧氧化,同時對葡萄糖代謝過程中活性氧的產生和清除具有調節作用。女性絕經后,卵巢功能衰退導致體內雌激素水平降低是AD發生的危險因素。雌激素缺乏可導致小鼠認知功能、葡萄糖攝取減少,葡萄糖轉運蛋白表達減少,線粒體能量代謝損害[30]。
5 結語
綜上所述, 葡萄糖攝取、利用和代謝調控障礙,與AD大腦中的能量合成障礙密切相關,但這三者之間的因果關系,特別是不同腦區、不同類型神經細胞中能量代謝障礙的差異目前還不十分明確。雖然體內參與能量合成的生物學過程相當復雜,但已有很多研究試圖從上述環節發現如何增強腦內葡萄糖攝取和利用,改善線粒體功能,提高大腦能量合成延緩AD的發生和進展,發現了許多改善能量合成障礙的化合物具有改善AD病理特征的作用,但大多數還處于動物實驗階段,為更好保證臨床應用的有效性和安全性,仍需進一步深入研究。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
人人健康(2023年26期)2023-12-07 03:55:46
學苑創造·A版(2020年9期)2020-10-13 09:41:02
中國生殖健康(2019年2期)2019-08-23 08:12:10
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
中國衛生標準管理(2015年1期)2016-01-14 03:41:27
藥學與臨床研究(2015年4期)2015-06-05 11:35:51
中國醫藥科學(2015年15期)2015-02-27 12:32:27
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
中國全科醫學(2013年36期)2013-01-25 06:20:58
