HPLC法測定斑蝥酸鈉注射液中斑蝥酸鈉的含量*
2021-09-25 09:05:34邢亞東羅凱文馬康李姍姍
河南中醫 2021年10期
關鍵詞:檢測
邢亞東,羅凱文,馬康,李姍姍
1.蚌埠醫學院,安徽 蚌埠 233030;2.悅康藥業集團安徽天然制藥有限公司,安徽 太和 236600
斑蝥酸鈉注射液收載于《化學藥品地方標準上升國家標準品種名單》中[1],屬于抗腫瘤藥,該藥可直接進入肝癌細胞核及核仁內,抑制癌細胞內DNA和RNA合成,影響癌細胞的核酸代謝,繼而使癌細胞形態和功能發生變化,殺死癌細胞,主要用于原發性肝癌、肺癌等癌癥的治療[2-4]。斑蝥酸鈉主要用于白細胞低下癥及肝炎、肝硬化、乙型肝炎[5-7],臨床應用較為廣泛。因斑蝥酸鈉注射液中斑蝥酸鈉的原標準檢測方法為氣相色譜法(gas chromatography,GC),具體過程為“以甲基硅膠(SE-30)為固定液,涂布濃度為10%;柱溫為180℃;氯仿溶解正十六烷,作為內標溶液。取斑蝥素對照品適量,精密稱定,加內標液適量,氯仿定容,作為對照品溶液。精密量取本品適量置錐形瓶中,加1 mol·L-1硫酸,氯仿萃取3次,每次10 min;萃取液合并,70℃水浴揮去氯仿至約5 mL,精密加內標液1 mL。混勻,轉移至10 mL量瓶中,用氯仿稀釋至刻度,搖勻,即為供試品溶液。測定,記錄色譜圖,以峰面積按內標法計算含量,再乘以1.316,即得。”[1]該過程使用的GC價格昂貴、操作較為繁瑣;樣品處理過程相對復雜、所用試劑毒性及危險性較大,在萃取過程中目標成分易損失,同時該檢測過程對檢測人員的整體素質要求較高。關于使用高效液相色譜法(high performance liquid chromatography,HPLC)檢測斑蝥酸鈉注射液中斑蝥酸鈉的含量無相關的報道[8-10]。為確保藥品在臨床使用中劑量更加精確以及藥品在生產過程中盡量減少對環境的污染,需要建立穩定、簡便、經濟的檢測方法控制該藥品的質量。現采用HPLC法定量測定斑蝥酸鈉注射液中斑蝥酸鈉的含量,可為斑蝥酸鈉注射液的質量控制提供參考依據。
1 材料
1.1藥物與試劑斑蝥酸鈉對照品(純度:99.0%,上海景榮化學科技有限公司,批號:20190930);斑蝥酸鈉維注射液(悅康藥業集團安徽天然 制 藥 有 限 公 司,批 號:19051201、19051202、19051203、19072101、19072102、19072103、19090601、19090602、19090603、19121401、19121402、19121403,規格:每支10 mL,每支含斑蝥酸鈉0.5 mg);甲醇、乙腈均為色譜純(TEDIA COMPANY,INC.),其余試劑均為分析純。
1.2儀器UltiMate3000型高效液相色譜儀(配置包括自動進樣器、柱溫箱、二極管陣列檢測器,美國賽默飛公司),GC2010plus氣相色譜儀(日本島津公司);BT125D型十萬分之一電子分析天平(德國賽多利斯公司);Esw Plus10型超純水機(上海茸研儀器有限公司);IKA RV 10 digital V型旋轉蒸發儀(IKA Germany);XZ133-SB-800D型高功率數控超聲波清洗器(北京中西遠大科技有限公司)。
2 方法
2.1色譜條件采用Phenomenex C18色譜柱(250.0 mm×4.6 mm,5 μm),流 動相為 乙腈-0.02 moL·L-1磷酸二氫鉀,等度洗脫,流速1.0 mL·min-1,柱溫30℃,進樣量10μL,檢測波長230 nm。
2.2溶液的制備
2.2.1 對照品溶液的制備 精密稱取斑蝥酸鈉對照品適量,加純水溶解并定容,制備成濃度為0.661 9 g·L-1的對照品溶液。
2.2.2 樣品溶液的制備 精密量取樣品溶液100 mL,60℃旋蒸濃縮至約5 mL,濃縮液轉移至10 mL棕色量瓶內并加水定容,即得。避光保存。
3 結果
3.1系統適應性實驗在室溫條件下,分別取斑蝥酸鈉對照品、供試品溶液適量,按色譜條件進樣,記錄色譜圖,見圖1。供試品和對照品溶液色譜圖相對應的位置上有相同的吸收峰。理論板數按斑蝥酸鈉計均大于6 000,斑蝥酸鈉與其他相鄰峰之間的分離度滿足ChP相關要求。
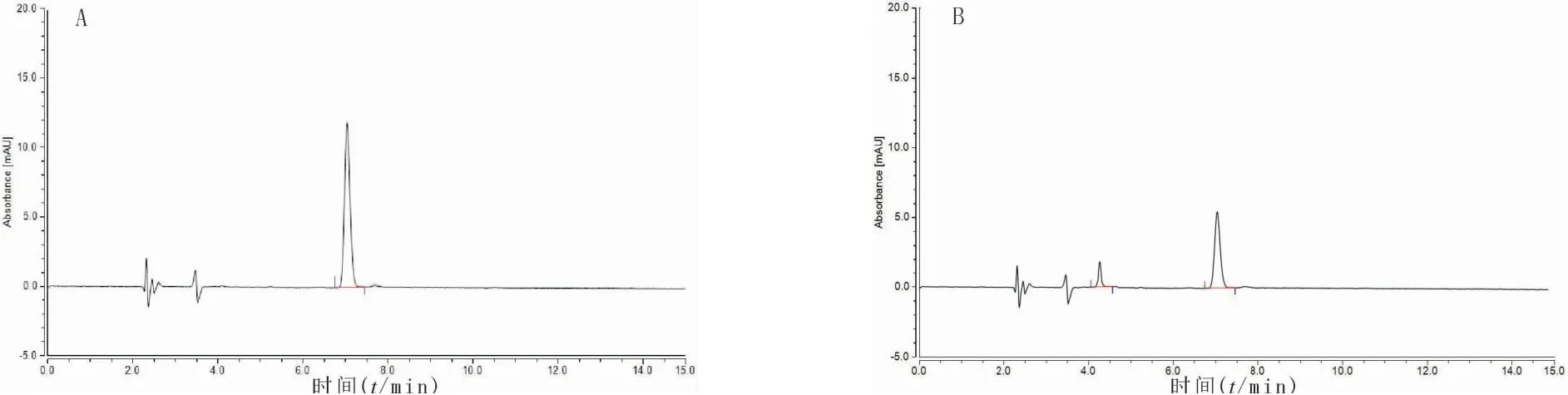
圖1 斑蝥酸鈉注射液色譜圖
3.2線性關系的考察分別精密量取“2.2”項下斑蝥酸鈉對照品溶液2μL、4μL、6μL、8μL、10μL注入高效液相色譜儀,記錄斑蝥酸鈉峰面積。以峰面積為縱坐標、進樣量(μg)為橫坐標進行線性回歸,得斑蝥酸鈉回歸方程分別為Y=0.753 3X+0.062 0(r=0.999 8)。結果表明斑蝥酸鈉在1.323 8~6.619 0μg范圍內線性關系良好。
3.3精密度實驗精密吸取濃度為0.661 9 g·L-1斑蝥酸鈉對照品溶液10μL,連續進樣6次,記錄斑蝥酸鈉峰面積,得出RSD值為0.64%,表明儀器精密度良好。
3.4穩定性實驗取同一供試品溶液(批號:19051201)適量,按色譜條件分別在0 h、1 h、3 h、6 h、15 h、24 h重復進樣一次,記錄斑蝥酸鈉峰面積,RSD值為1.17%。結果表明斑蝥酸鈉在該條件下24 h內穩定。
3.5重復性實驗取同一批號(批號:19051201)供試品適量,按樣品制備方法平行制備供試品溶液6份。按色譜條件進樣分析,記錄斑蝥酸鈉峰面積,RSD值為0.83%。結果表明方法重復性良好。
3.6加樣回收率實驗取已知含量的斑蝥酸鈉注射液(批號:19051201)樣品15份,分3組,每份約50 mL,精密量取,置于100 mL量瓶中,再分別精密加入一定量的斑蝥酸鈉對照溶液(分別相當于含量的80%、100%、120%的量)并加水定容,按“2.2.2”項下方法制備供試品溶液,再按上述測定含量的方法測定,記錄峰面積,計算得斑蝥酸鈉的平均回收率為98.51%,RSD為0.96%(n=15)。結果表明該法加樣回收率良好。見表1。
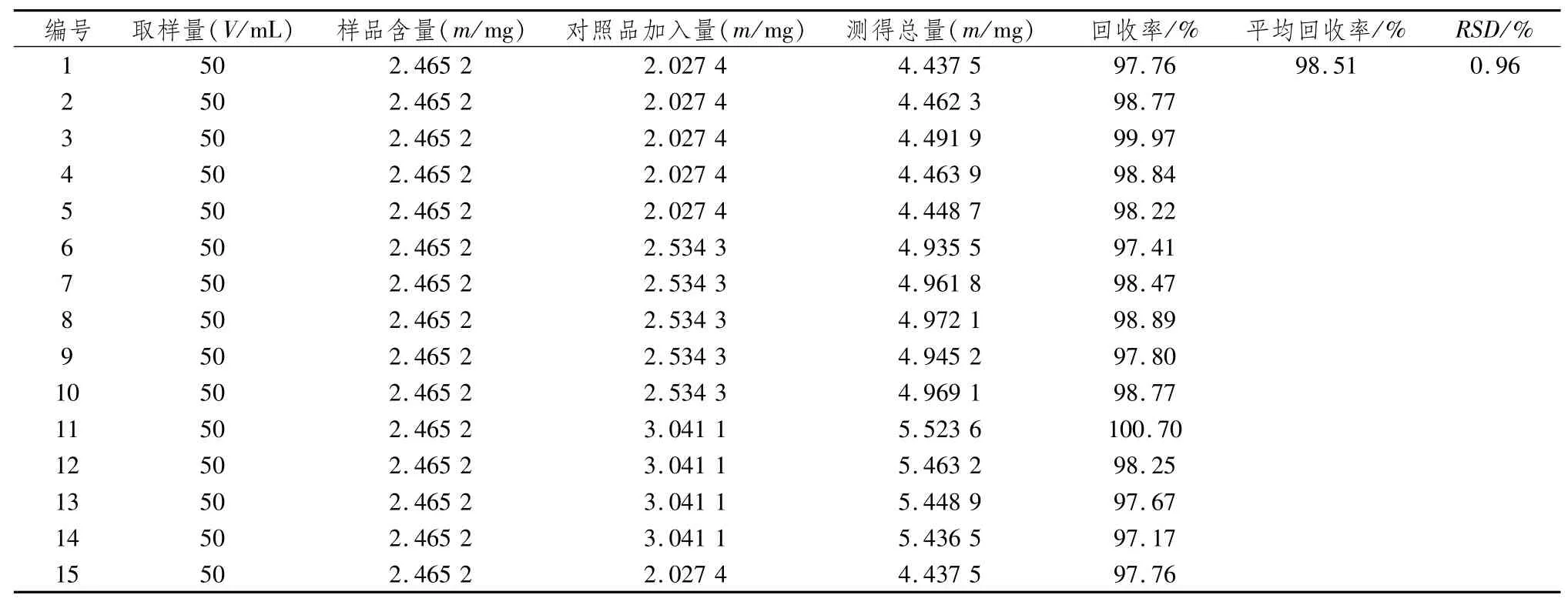
表1 斑蝥酸鈉注射液中斑蝥酸鈉加樣回收率實驗結果 (n=15)
3.7樣品含量測定取收集的12批斑蝥酸鈉注射液適量,精密量取適量,按“2.2.2”項下方法制備供試品溶液,按相應色譜條件進樣測定并記錄斑蝥酸鈉峰面積,計算斑蝥酸鈉含量,同時按國家相應的原標準檢測方法(GC法)對12批樣品中斑蝥酸鈉的含量進行測定,結果見表2。
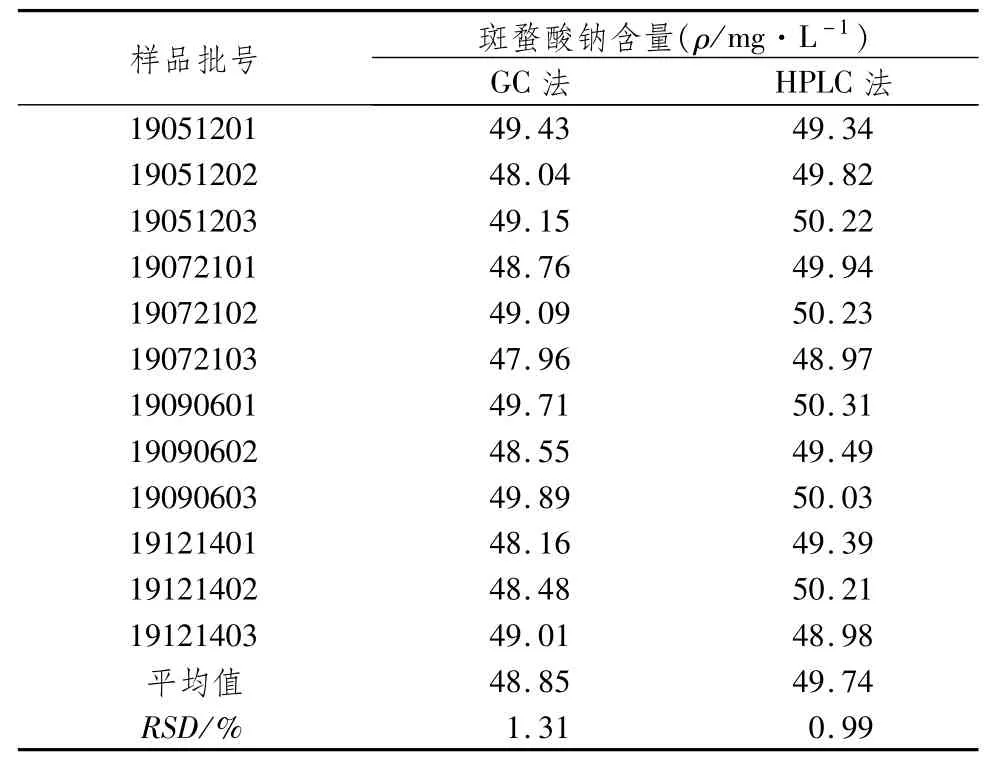
表2 含量測定結果 (n=3)
4 討論
斑蝥酸鈉為斑蝥素的衍生物,與斑蝥素比較,其抗腫瘤療效確切,不良反應較少[11-13],可有效改善放、化療的不良反應,提高腫瘤患者的免疫功能及生存質量,為臨床常用藥[14-16]。但斑蝥酸鈉注射液收載的檢測方法為GC法,該法制備樣品較為復雜,目標成分易損失,而其他的檢測方法報道較少。針對上述存在的問題,本實驗建立了HPLC法測定斑蝥酸鈉注射液中斑蝥酸鈉的含量,并對所建立的相關實驗條件進行摸索與優化,從而為其質量控制提供參考依據。
對斑蝥酸鈉在紫外條件下進行全波長掃描,發現其在193 nm和230 nm處有最大吸收,對樣品使用193 nm波長進行含量測定時,基線波動不利于整個測定過程的順利進行,而230 nm波長時基線較穩,更有利于檢測過程的進行。因此,選擇230 nm作為檢測波長。
采用HPLC法測定斑蝥酸鈉的文獻較少,故本次實驗分別摸索了甲醇、乙腈-水[17-20];乙腈-0.02 moL·L-1磷 酸 二 氫 鉀 溶 液[21-24];乙 腈-0.1%磷酸(三乙胺調pH為2.80)[25-28];乙腈-0.1%磷酸水溶液[29-30];乙腈-0.005 moL·L-1辛烷磺酸鈉溶液(用H3PO4調pH為2.50)等流動相體系[31]。結果發現,含甲醇的流動相體系對實驗有干擾,原因可能為甲醇存在末端吸收,斑蝥酸鈉在乙腈-0.1%磷酸(三乙胺調pH為2.80);乙腈-0.1%磷酸水溶液;乙腈-0.005 moL·L-1辛烷磺酸鈉溶液體系中峰型的對稱度較差,拖尾嚴重;且辛烷磺酸鈉屬于大分子物質,在流動相的制備和使用過程中容易產生氣泡,不利于整個實驗的進行和儀器的保護,在乙腈-0.02 moL·L-1磷酸二氫鉀溶液體系中保留時間較為合適、峰型較好、柱效較高,有利于整個檢測實驗的順利進行。
本實驗根據樣品的實際情況建立的HPLC法測定斑蝥酸鈉注射液中斑蝥酸鈉含量只需對樣品溶液進行簡單的濃縮、定容,樣品處理過程簡單、方便、易操作,儀器使用簡便,且測定結果較為穩定。所建立的方法與原標準中收載的方法相比存在一定的優勢,該方法可用于對斑蝥酸鈉注射液質量進行控制,為進一步改進斑蝥酸鈉的檢測方法奠定了一定的基礎。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
