生物油電催化加氫提質技術研究進展
2021-10-31 23:36:06鄧偉林鎮(zhèn)浩熊哲汪雪棚徐俊江龍蘇勝汪一胡松向軍
化工學報 2021年10期
鄧偉,林鎮(zhèn)浩,熊哲,汪雪棚,徐俊,江龍,蘇勝,汪一,胡松,向軍
(1華中科技大學煤燃燒國家重點實驗室,湖北武漢430074;2香港城市大學能源與環(huán)境學院,香港999077)
引 言
化石能源緊缺和日益嚴峻的環(huán)境挑戰(zhàn)正在推動世界各國積極開發(fā)清潔可再生新能源。在“2060碳中和”目標背景下,我國明確將加大力度大規(guī)模發(fā)展清潔能源。生物質作為僅次于煤、石油、天然氣的第四大能源,具有碳中和特點,生物質的大規(guī)模利用有助于實現(xiàn)“碳中和”目標。目前,可大規(guī)模利用的生物質主要指農業(yè)和林業(yè)廢棄物。我國擁有極為豐富的農林廢棄物資源,每年可利用的農林廢棄物資源產量約20億噸[1-3]。然而,由于農林廢棄物本身存在比體積大、季節(jié)性強及分散度高等固有特點,導致其收集難、儲運難、利用效益低,制約了農林廢棄物類生物質資源化、清潔化利用。而熱解多聯(lián)產技術可將農林廢棄物高效、連續(xù)轉化為熱解氣、生物炭、生物油三種高附加值產品,契合我國農林廢棄物數(shù)量大、密度低、分散度高等特征屬性,具有良好的推廣應用前景[4-8]。
生物油是生物質快速熱解的主要產物,是唯一以液體形式存在的含碳可再生能源,具備替代化石能源的潛力。然而,不同于熱解氣與生物炭可分別作為燃料和肥料緩釋載體直接高值化利用,生物油至今仍未有成熟可工業(yè)化的應用或處理技術,成為制約熱解多聯(lián)產技術路線提高其綜合效益的瓶頸。導致生物油的難應用、難處理的根本原因是其特有的物理化學特性。原始生物油含氧量高、熱值低,必須進行精制提質以提升其燃料性質。近年來,國內外研究人員探索了生物油提質技術,研究主要集中于加氫脫氧、催化裂解、水蒸氣重整等傳統(tǒng)熱化學方法[9-12]。然而,生物油中存在大量含氧基團,在高壓高溫條件下極易聚合產生積炭,造成反應器堵塞和催化劑失活,反應難以持續(xù)。為此,國內外學者開始積極探索溫和條件下的生物油提質方法,如電催化加氫[13-14]等。圖1顯示了近十年來以生物油電催化提質為主題的論文數(shù)量逐年增長,說明電化學催化加氫提質受到越來越多的關注。
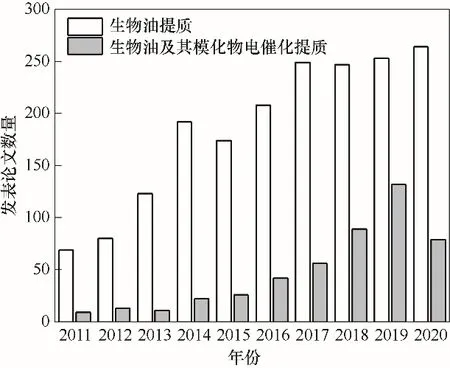
圖1 近十年發(fā)表的生物油提質及生物油電催化提質相關論文(來源:Web of Science)Fig.1 Annual published papers related to bio-oil upgrading and bio-oil electrocatalytic upgrading in the last decade(Source:Web of Science)
電催化加氫提質反應可在常溫常壓下進行,無須外部氫氣供應,工藝簡單;通過調節(jié)電位即可快速實現(xiàn)反應啟停和產物選擇性調控,運行簡便;系統(tǒng)規(guī)模靈活,便于組成分布式系統(tǒng);可利用可再生能源(太陽能、風能)提供電力,實現(xiàn)全過程碳中和。電催化提質還有助于減少溫室氣體排放[15]。研究表明,電催化加氫可將生物油模型化合物轉化為含氧量低、酸度低的產物[16-20],還可降低生物油水溶性組分酸度,提高其化學穩(wěn)定性[21-22]。可見,電催化加氫在生物油提質中具有顯著潛力。
本文綜述了近年來生物油電催化加氫提質的研究進展,分析了典型的生物油模型化合物在電催化加氫過程中的反應機理,討論了原始生物油電催化加氫提質的不同實例。最后闡述了生物油電催化加氫提質技術面臨的困難和挑戰(zhàn),提出了未來該領域的研究重點和方向。
1 生物油特性及提質技術
生物質通過快速熱解可轉化為生物油,同時還產生少量的熱解氣和生物炭。生物油的實際組成隨原料和熱解條件的變化而有所不同,已有研究總結了生物油的一般特性[23-26]。首先,新制得的生物油是一種由15%~30%(質量分數(shù))的水和大量含氧有機化合物組成的酸性深色糖漿似的乳狀液。其次,生物油的低位熱值為13~18 MJ/kg,遠低于車用汽油的46 MJ/kg,生物油熱值較低的原因在于其水分多、氧含量高。生物油的碳含量通常約為50%(質量分數(shù)),硫和氮含量較低。生物油中含有甲酸和乙酸,其pH在2.5左右,具有腐蝕性。生物油含有300多種不同的化合物,其中約有20%(質量分數(shù))的成分由于其高分子量和復雜的化學結構而無法用氣相色譜-質譜儀(GC-MS)或液相色譜-質譜儀(LC-MS)檢測到[27]。生物油中可用GC-MS檢測的化合物包括C2~C4含氧烯烴,C2~C6含氧有機物,如醛、酮、羧酸、(脫水)糖、呋喃,以及大量C6~C8甲氧基化芳香族化合物,如表1所示[28-31]。生物油中大量高度活潑的含氧官能團導致其熱穩(wěn)定性差,化合物之間的二次反應導致生物油老化,黏度隨儲存時間而增大。羰基官能團和羧基官能團的組合使得原始生物油在儲存過程中極易聚合[8,32]。相關模型化合物研究表明,糖、酸、呋喃和芳香組分也會促進聚合過程[33-34]。因此,提高生物油的穩(wěn)定性成為提高其可利用性的關鍵,以便將其儲存或運輸?shù)郊袩捰蛷S進行進一步提質。
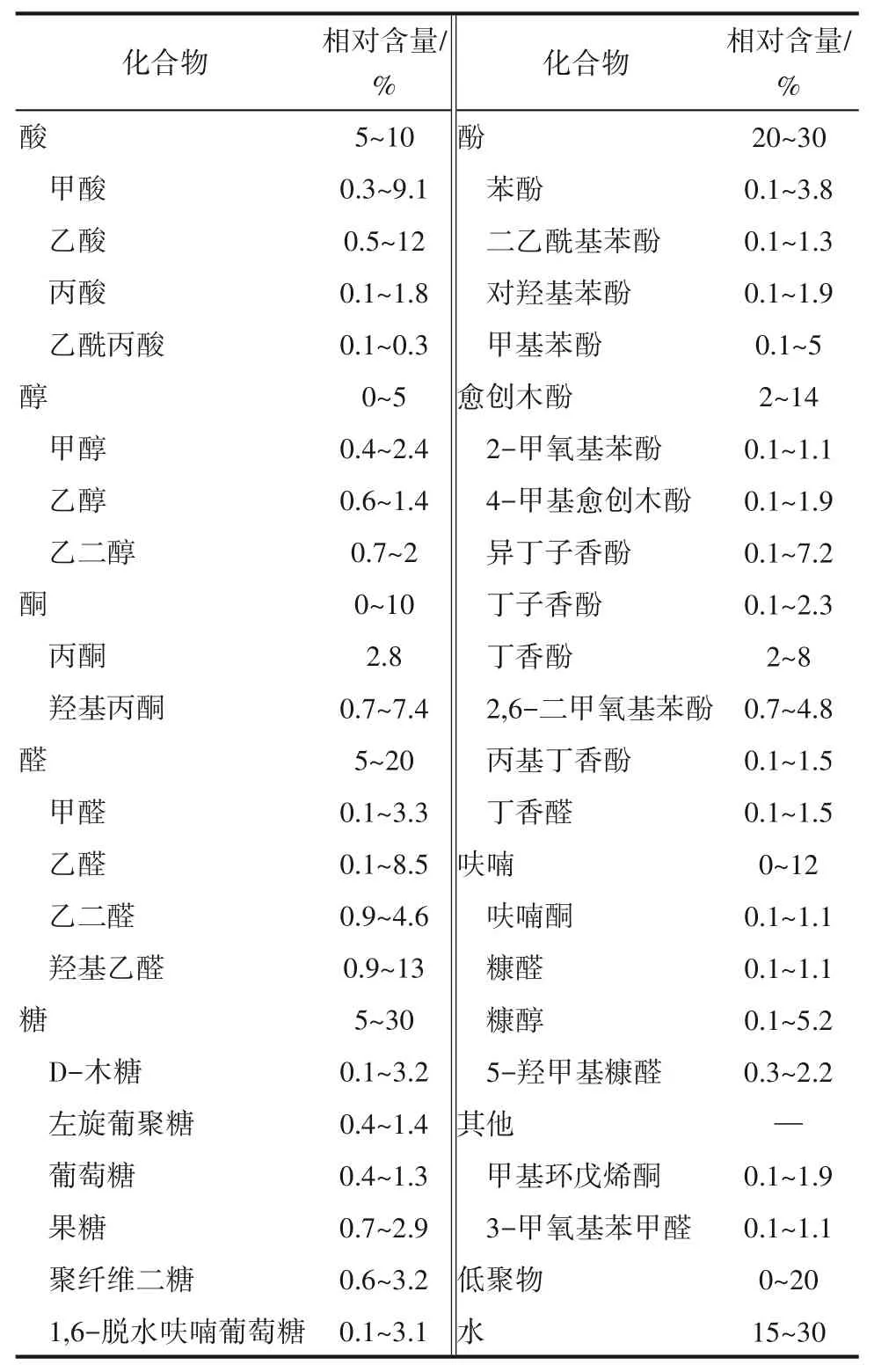
表1 生物油的一般組成Table 1 General composition of bio-oils
生物油成分復雜、含氧量高、熱值低、酸值高、黏度大、穩(wěn)定性差的特點導致生物油難應用、難處理,必須進行精制提質以使其能夠穩(wěn)定儲存和運輸,從而擴大生物油的應用領域并提升其利用價值。一直以來,國內外研究人員探索了各種提質策略來改善原始生物油的腐蝕性、化學不穩(wěn)定性和較低的熱值[35-43]。表2歸納了不同的生物油提質方法。
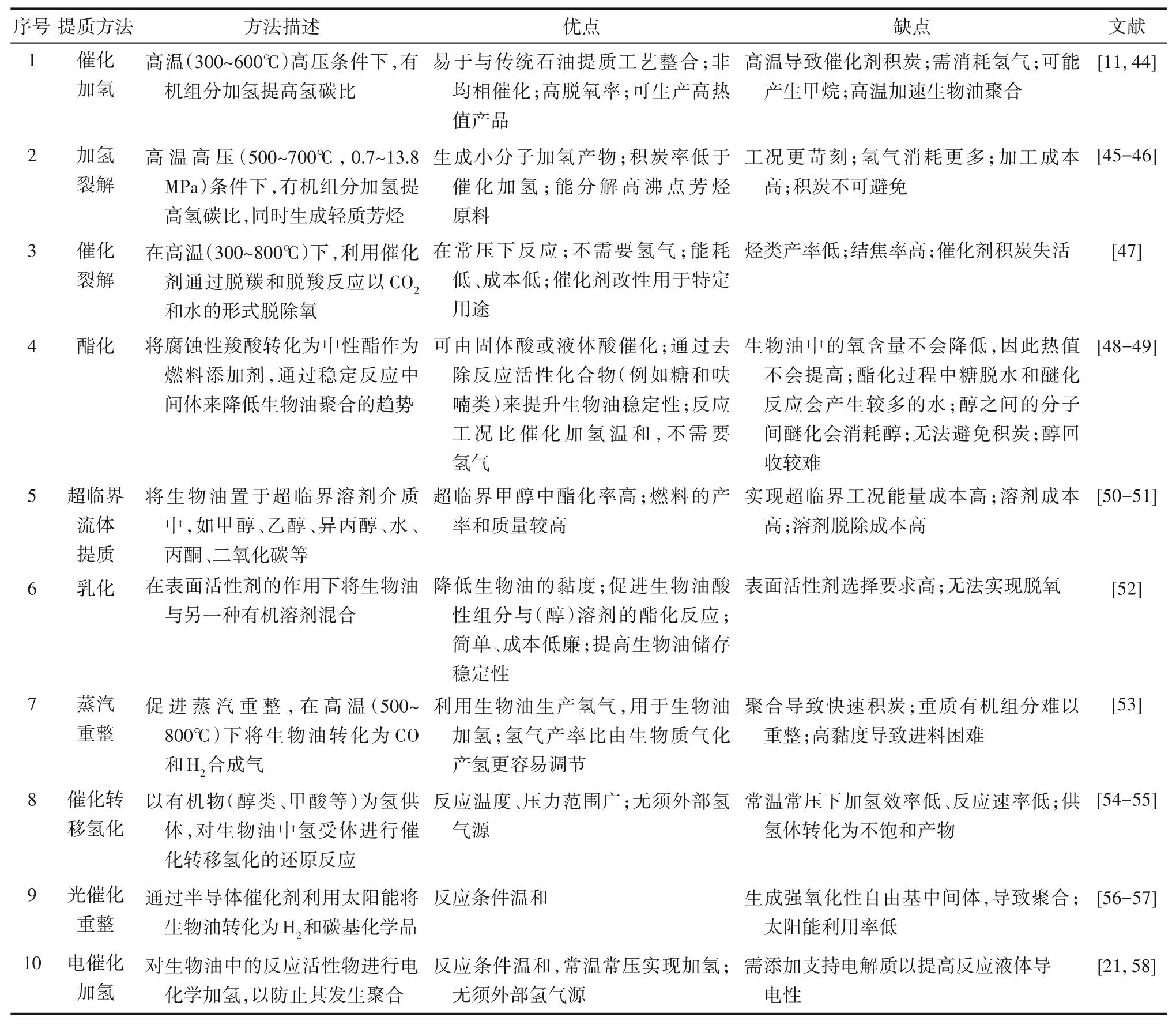
表2 生物油提質技術Table 2 Summary of bio-oil upgrading techniques
2 電催化加氫提質
電催化加氫提質方法的優(yōu)勢在于它能夠在常壓下低于100℃的溫和條件下利用小電位差實現(xiàn)加氫。電催化加氫反應所需氫源可通過質子性溶劑的電離來提供,因此無須外加氫氣源。可見,電催化加氫避免了高溫高壓加氫處理所面臨的反應條件嚴苛、氫氣消耗大、成本高等問題。除了溫和的反應性質外,電催化反應裝置簡單,可在未分隔電解池或由質子交換膜分隔的H型雙腔室電解池中進行,當加氫產物不會被陽極氧化時可優(yōu)先使用未分隔電解池。對于H型電解池,電催化反應過程可以被分成不同的半反應,包括陰極還原和陽極氧化,每個反應獨立進行,而熱化學過程中氧化還原反應很難區(qū)分開。
圖2顯示了加氫反應的基本原理。在陰極室中,原始生物油的電催化加氫反應[式(1)]和析氫反應[式(2)]發(fā)生在陰極表面;在陽極室中,水[式(3)]或有機基質[式(4)]的氧化在陽極表面發(fā)生,并產生陰極室生物油加氫反應所需要的氫和電子。
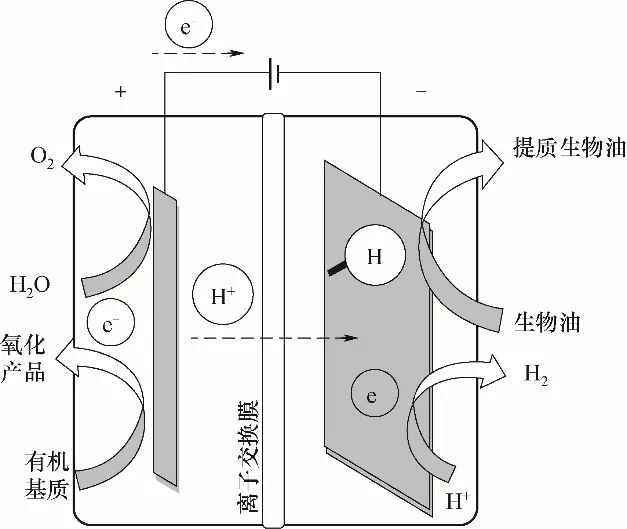
圖2 生物油電催化加氫原理示意圖Fig.2 Schematic diagram of principle of electrocatalytic hydrogenation of bio-oil
陰極:
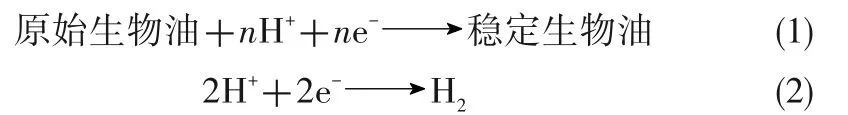
陽極:

由此可見,生物油電催化加氫包括如下步驟:原子氫生成(nH++ne-)和陰極吸附、生物油有機化合物的陰極吸附、加氫反應以及加氫產物脫附。電催化反應的本質可理解為質子和電子協(xié)同轉移的能力。電子轉移一般通過如下兩種形式實現(xiàn)。
(1)直接轉移,即反應物吸附在電極表面,電子轉移直接發(fā)生在固液界面,反應物被直接氧化或還原。
(2)間接轉移,即反應介導物先在電極表面通過得失電子生成活性中間體,中間體擴散到液相中與反應物發(fā)生電子傳遞產生氧化還原產物。生物油中含有大分子有機物,較大的分子尺寸一定程度上阻礙其在電極表面吸附,因此生物油電催化加氫反應過程更有可能遵循電子間接轉移模式。生物油加氫反應[式(1)]和析氫反應[式(2)]是競爭反應。一般用法拉第效率(FE)即電流效率(CE)對電催化加氫性能進行評估。
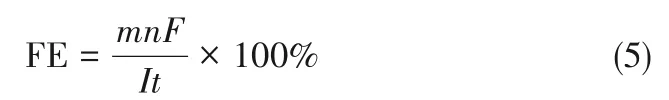
式中,m為產物量,mol;n為生成產物所消耗的電子數(shù);F為法拉第常數(shù),96485 C/mol;I為反應電流,A;t為反應時間,s。
由于法拉第效率與多個實驗參數(shù)(溫度、電位、pH、反應物濃度、催化劑)相關,很難提供使其優(yōu)化的一般準則。因此,法拉第效率的優(yōu)化通常采取試錯法。通過改變實驗參數(shù),監(jiān)測目標產物產率的響應。然而,在生物油電催化加氫中,由于難以對特定產物進行定量,通常首先利用生物油模型化合物對實驗參數(shù)進行優(yōu)化,以獲得最高的法拉第效率,然后將優(yōu)化的實驗條件應用于生物油的提質。
3 生物油模型化合物電催化加氫反應機理
生物油模型化合物電催化加氫的研究備受關注。研究體系主要以單種模型化合物為主,涵蓋生物油主要低分子化合物。本節(jié)以化學官能團劃分模型化合物,分別評述各類模型化合物的電催化加氫反應機理研究進展。
3.1 醛酮類化合物
小分子醛和酮是導致生物油化學性質不穩(wěn)定的主要原因之一。這些羰基化合物是由纖維素和半纖維素在快速熱解過程中降解生成的。盡管生物油組分在不同的原料和熱解條件下有所不同,但醛和酮(如羥丙酮、丁醛、糠醛和環(huán)戊酮等)一直是生物油的主要組成部分。Black等[59]測定的橡木生物油的羰基含量接近4 mol/kg。在酸性環(huán)境下,羰基化合物容易聚合,影響生物油的黏度和化學成分。此外,酚類和小分子醛也容易發(fā)生聚合。因此,羰基的電催化加氫是提高生物油化學穩(wěn)定性的關鍵。羰基加氫會轉化為醇或二醇[22],如圖3所示。
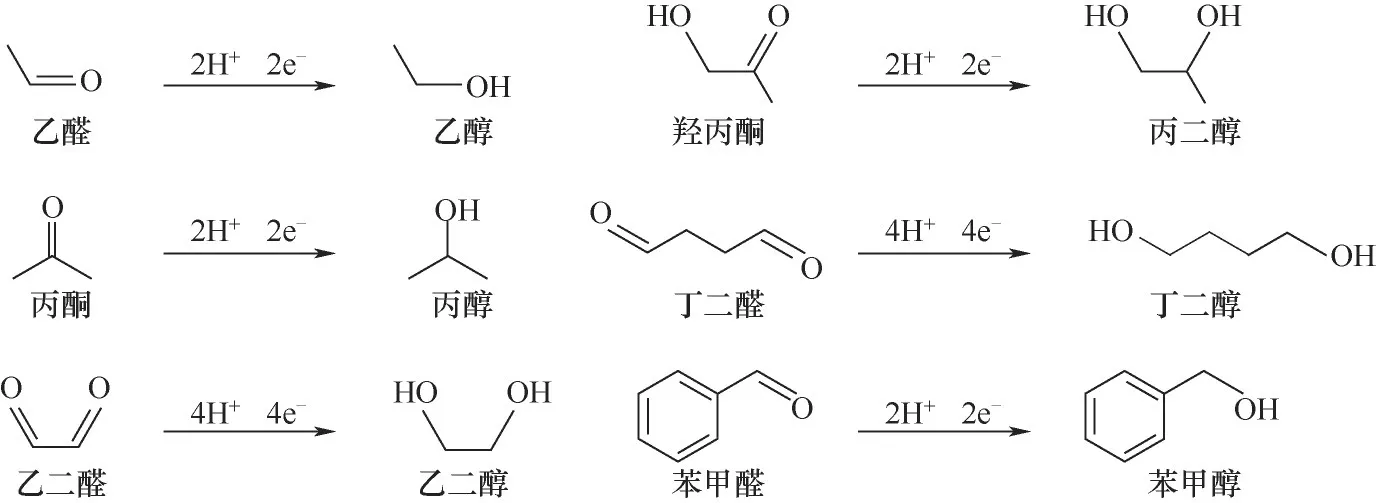
圖3 醛、酮等羰基化合物的電催化加氫反應機理Fig.3 Reaction mechanism of electrocatalytic hydrogenation of carbonyl compounds
各種貴金屬和非貴金屬催化劑電催化加氫羰基化合物的性能被廣泛研究。盡管貴金屬被普遍認為具有優(yōu)異的催化性能,但某些非貴金屬在電催化加氫羰基化合物上表現(xiàn)出更好的性能[60]。例如,在羥基丙酮轉化為丙二醇[61]以及糠醛轉化為糠醇[62]的反應中,Ni的活性高于Pt。貴金屬的低加氫活性通常歸結于析氫競爭反應,析氫阻礙了有機反應物的吸附。Andrews等[63]研究了一系列非貴金屬(Cu、Ni、Co)和貴金屬(Pd、Ru、Rh)在不同脂肪族和芳香族羰基化合物上的電催化加氫性能。結果表明,Cu和Pd在苯甲醛加氫制備苯甲醇過程中具有較高的活性;而Ni、Zn和Co在加氫形成苯甲醇時活性較低,同時生成二聚產物氫化苯偶姻。此外,該研究也指出反應物和電極表面間強大的結合能能夠顯著促進反應物加氫[64]。在生成共軛羥基或存在強酸的情況下,羰基化合物會脫水生成烯烴,然后加氫生成烷烴。從能量的角度來看,脫羥基(脫水)是有益的,因為脫羥基去除了氧原子,從而增加了分子的能量密度。然而,脫羥基產物由于低沸點更容易蒸發(fā),因此,需通過使用冷阱或溶劑捕集阱收集疏水產物。
3.2 有機酸類化合物
有機酸(如甲酸、乙酸和丙酸)在典型的電催化加氫條件下(低于100℃,0.1MPa)幾乎不發(fā)生還原反應,因此有機酸在電催化加氫反應體系中受到的關注相對較少。Cirtiu等[65]研究發(fā)現(xiàn)乙酸、丙酸、丁酸等脂肪酸有利于促進有機物在催化劑表面的吸附和解吸,從而提高了其他組分(如苯酚)的電催化加氫速率。然而,脂肪酸本身是否也發(fā)生了加氫并沒有被提及。盡管有機酸到醇或烷烴的轉化仍然很困難,但還有其他方法來中和生物油的酸性。一種廉價簡易的解決方案是利用堿溶液[28]或堿廢料中和酸度[66]。通過電透析方法也可以將生物油中的酸組分分離出來[21]。
相比之下,羰基的電催化加氫反應性遠比羧酸基團更強。以乙酰丙酸的電催化加氫為例,乙酰丙酸中的羰基先經(jīng)過加氫還原成羥基,生成4-羥基戊酸,然后仲羥基經(jīng)過脫水生成戊酸。與乙酸相比,戊酸具有更大的燃料潛力,它可以通過Kolbe反應氧化生成辛烷。因此可通過簡便的兩步反應從生物質衍生的乙酰丙酸中獲取辛烷[67]。戊酸和γ-戊內酯均可在Pb電極上生成,其選擇性可通過過電位和電解液pH來調節(jié)。較低的過電位和較高的pH(-1.3 V(vsRHE),pH 7.5)有利于γ-戊內酯的生成,其選擇性達100%;而較高的過電位和較低的pH(-1.5 V(vsRHE),pH 0)則有利于戊酸的生成,其選擇性達97%[68],如圖4所示。
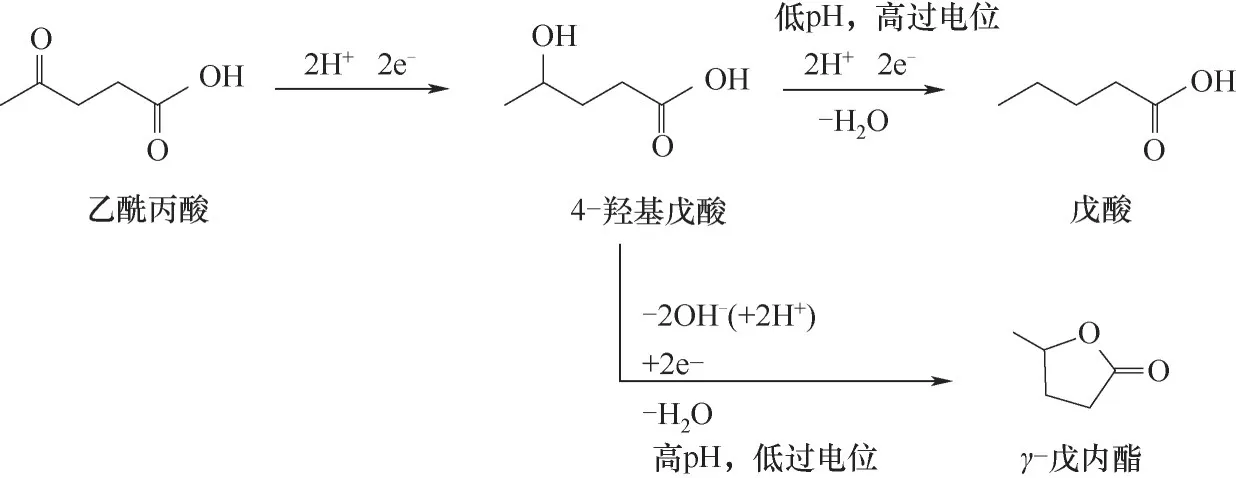
圖4 乙酰丙酸在不同電位和pH條件下的電催化加氫反應機理Fig.4 Reaction mechanism of electrocatalytic hydrogenation of levulinic acid at different potentials and pH conditions
羰基和羧酸基團的反應性差異突出了電催化加氫在生物油提質中的適用性,即大量羧酸不會阻礙羰基化合物的加氫反應。在理想情況下,將羧酸轉化為醇或烷烴能提高生物油的燃料價值。其他與生物油相關的有機酸,如香草酸和丁香酸的電催化加氫尚未研究。當然這些有機酸含量少,酸性比乙酸弱,對生物油的化學穩(wěn)定性的影響較小[69]。
3.3 糖類化合物
生物油中含有多種由纖維素和半纖維素解聚而成的單糖。葡萄糖的電催化加氫可生產食品添加劑或其他高值化學品(如維生素C)前體。Kwon等[70]研究了中性環(huán)境下葡萄糖在30種不同金屬電極上的加氫反應,發(fā)現(xiàn)產物的選擇性主要取決于催化劑種類,而電位和pH對其影響較小。催化劑按產物分布被分為三類(圖5):(1)Ni、Fe、Co、Cu、Pd、Au和Ag有利于山梨醇的形成;(2)Pb、Zn、Cd、Sn、In、Sb和Bi有利于山梨醇和2-脫氧山梨醇的形成;(3)Ti、V、Cr、Mn、Zr、Nb、Mo、Hf、Ta、We、Re、Ru、Rh、Ir和Pt有利于析氫反應。其中,2-脫氧山梨醇不是在電極表面直接催化脫羥基,而是通過脫水反應生成。
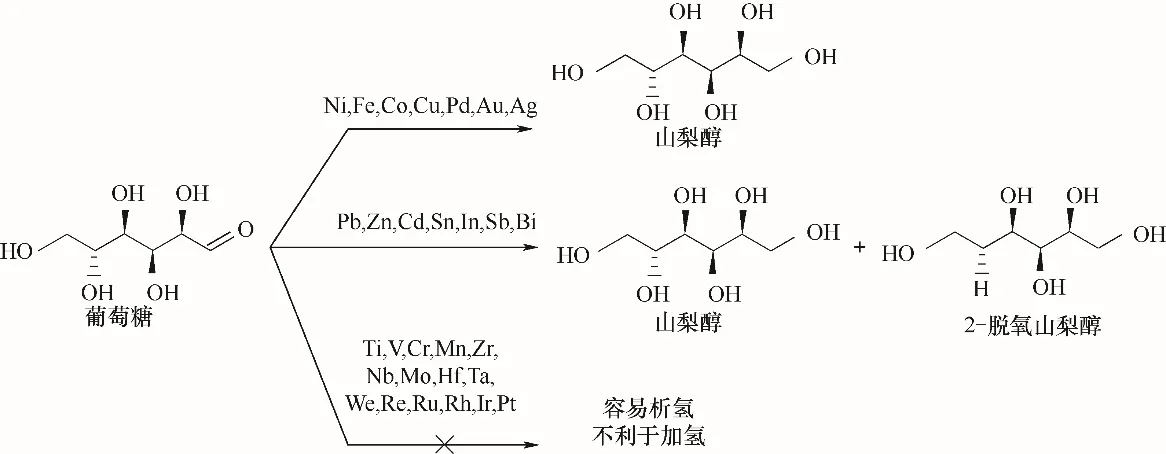
圖5 葡萄糖在不同金屬電極作用下的電催化加氫反應機理Fig.5 Reaction mechanism of electrocatalytic hydrogenation of glucose with different metal electrodes
3.4 呋喃類化合物
糠醛可在電催化加氫體系下生產高值化燃料和化工產品[16,18,71-73]。生物油中糠醛的濃度隨生物質原料不同而異,且與半纖維素的含量有關[74-75]。糠醛的加氫產物主要是糠醇(FA)和2-甲基呋喃(MF)。如圖6所示,在電催化加氫反應中,糠醛先通過羰基還原轉化為糠醇,糠醇通過脫水反應生成2-甲基呋喃。兩種電催化加氫產物的選擇性控制機理得到廣泛研究[62,76-79]。然而,在高酸性環(huán)境中,糠醇和2-甲基呋喃都可能發(fā)生聚合副反應,導致目標產物的產率降低。與糠醇相比,2-甲基呋喃的沸點更低,疏水性更強,可能在反應中揮發(fā)[76]。
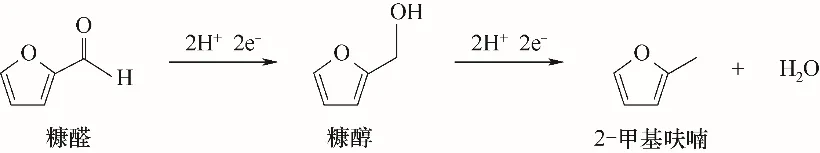
圖6 糠醛電催化加氫反應機理Fig.6 Reaction mechanism of electrocatalytic hydrogenation of furfural
Li等[80]在對糠醛的電催化加氫研究中提出了一種收集疏水產物的簡便方法,即采用辛烷溶劑捕集阱,將產物產率從64%提高到85%。然而,糠醛也部分溶解在辛烷溶劑中,一定程度上降低了有效反應物的含量。該研究中還發(fā)現(xiàn),通過Ni犧牲陽極提供了一個恒定的鎳離子流,在陰極上采用電沉積的方式使陰極表面再生,可以促進糠醛的加氫反應。此外,該研究還比較了幾種不同陰極電極材料的糠醇轉化率,轉化率從大到小依次為Fe、Ni、Cu、308不銹鋼和Al。
Chadderdon等[78]研究發(fā)現(xiàn)糠醛還原的第一個電子轉移發(fā)生在電極表面的外層,隨后消耗一個吸附的氫產生糠醇和部分2-甲基呋喃。May等[81]以Cu為陰極,總結了pH、過電位和反應時間對糠醛電催化加氫合成糠醇或2-甲基呋喃的影響機制。研究表明,pH在3~5范圍內,糠醇具有較高的選擇性;而pH低于2時,有利于2-甲基呋喃的生成。在0.5 mol/L H2SO4中,工作電位保持在500 mV和650 mV(vsRHE)左右時,可有效將糠醛轉化為糠醇和2-甲基呋喃,同時最大限度抑制析氫副反應。研究還建議糠醛的初始濃度應在20~100 mmol/L之間,以避免形成低聚物。溶劑組成和反應時間對產物選擇性的影響有限[81]。這些研究結果證明了電催化加氫適用于提質生物油中的呋喃類化合物,因為生物油的pH約為2.5,非常接近有利于形成2-甲基呋喃的pH,同時生物油中糠醛含量一般為0.1%~1.1%(相當于10.4~114 mmol/L,如表1所示),這也與推薦的糠醛濃度范圍相當接近。
5-羥甲基糠醛(HMF)是生物油中另一種典型呋喃類化合物。Kwon等[82]分別對HMF、葡萄糖以及二者混合組分在中性電解液中進行了電催化加氫實驗。該研究篩選了一系列金屬電催化劑,并且按照產物選擇性分為三組:(1)Fe、Ni、Ag、Zn和Cd,主要產物為2,5-二羥甲基呋喃(DHMF);(2)Pd、Al、Bi和Pb,生成DHMF和其他氫解產物,產物分布受電位的影響;(3)Co、Au、Sn和Sb,主要產物為氫解產物。這些催化劑表現(xiàn)出幾乎相同的起始電位,說明引發(fā)HMF加氫反應與催化劑種類關聯(lián)較小,但催化劑的內在性質會影響產物分布。同時,研究還發(fā)現(xiàn)在Fe、Ni、Ag、Pd、Co、Au和Cu上進行電催化加氫反應時,葡萄糖的存在會促進HMF加氫轉化為DHMF,而HMF會抑制葡萄糖加氫產物山梨醇的生成。Kwon等[83]又在酸性電解液中用上述電催化劑進行了HMF加氫反應研究,發(fā)現(xiàn)在Cu、Ni、Fe和Pb電催化劑作用下主要產物為DHMF,而Pt、Pd、Al、Zn、Sb和In主要產物為2,5-二羥甲基四氫呋喃(DHMTHF),如圖7所示。研究發(fā)現(xiàn)酸性環(huán)境可降低HMF的氫過電位,并促進其轉化為不同的加氫產物。這兩項研究都驗證了電催化加氫作為生物油提質策略的可行性。生物油的高酸度以及富含葡萄糖的特性都有助于HMF通過電催化加氫轉化為飽和產物。
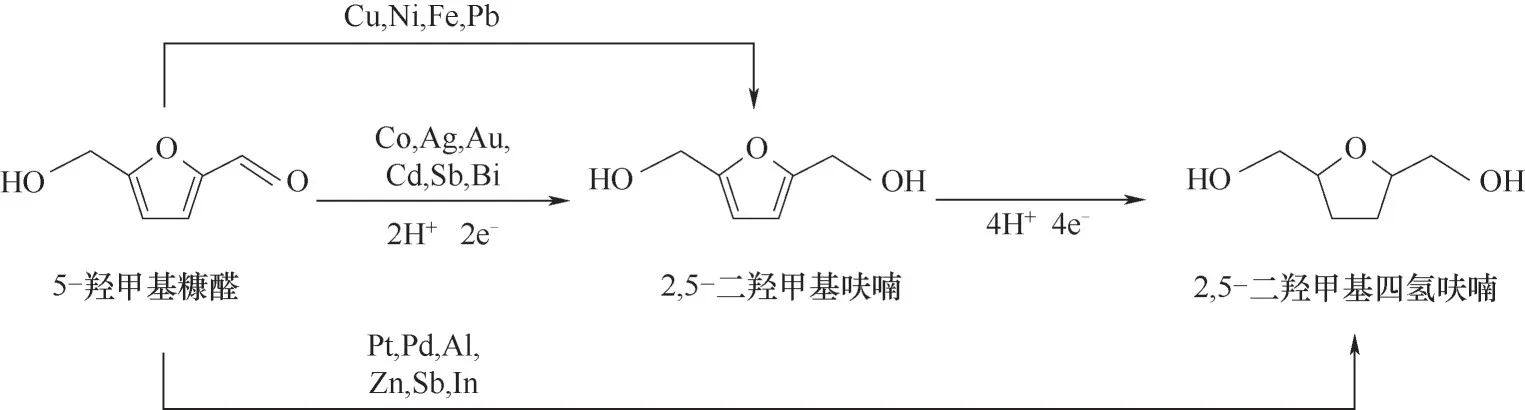
圖7 HMF在不同金屬電極作用下的電催化加氫反應機理Fig.7 Reaction mechanism of electrocatalytic hydrogenation of HMF with different metal electrodes
3.5 芳香族化合物
芳香族化合物是由木質素在熱解過程中降解產生的,是生物油的重要組分之一[19,84]。生物油中常見的芳香族化合物包括苯酚、愈創(chuàng)木酚、二甲氧基苯酚(紫丁香醇)等。目前,芳香π鍵的電催化加氫僅在貴金屬催化劑(如Pt[85-86]、Rh[87-89]和Ru[90-91])以及多孔結構金屬催化劑(如Raney-nickel[92-94])上實現(xiàn)。Pd也可以實現(xiàn)芳香π鍵的電催化加氫[65,93,95],但它對芳香π鍵的電催化加氫催化活性較低[88]。
Liu等[86]采用碳載Pt(Pt/C)電催化劑與硅鎢酸(SiW12)組成“雙催化劑體系”對一系列生物油衍生芳香族單體化合物進行了電催化加氫實驗,發(fā)現(xiàn)大多數(shù)產物的轉化率可達90%以上,主要產物為環(huán)脂肪醇,其中一部分經(jīng)過酸催化脫水轉化為飽和烷烴。Zhou等[96]通過有序介孔碳負載硼摻雜PtNi合金的方式制備了復合納米材料電催化劑(PtNiB/CMK-3),不僅成功將木質素單體轉化為環(huán)己醇和環(huán)己酮,實現(xiàn)86.2%的法拉第效率和97.5%的轉化率,同時該催化劑還表現(xiàn)出很強的穩(wěn)定性,負載的PtNi納米粒子在反應前后不易溢出基底或團聚,顯示出了優(yōu)良的加氫性能。
愈創(chuàng)木酚的電催化加氫可能存在兩種不同的反應路徑:(1)芳香環(huán)直接加氫生成甲氧基環(huán)己醇;(2)芳基C—O鍵氫解生成苯酚,隨后加氫生成環(huán)己醇。芳基C—O鍵的斷裂從愈創(chuàng)木酚中除去一個氧原子,從而提高其熱值。通過總結若干電催化脫甲氧基反應的研究[87-88,90-91,93,96-98],可概括如下:(1)無論電極材料如何,C—O鍵的斷裂只優(yōu)先發(fā)生在芳基結合位點而不是苯環(huán)側鏈中的烷基位點,即不會產生鄰苯二酚及其衍生物;(2)芳香環(huán)π鍵加氫反應對催化劑的敏感性不高,且低電流密度下更有利于脫甲氧基反應[90,94];(3)脫甲氧基反應只發(fā)生在芳香環(huán)加氫過程中,而脂肪族醇(如2-甲氧基環(huán)己醇)不能直接脫甲氧基生成環(huán)己醇(圖8)。
Lam等[94]研究了愈創(chuàng)木酚等甲氧基芳香族化合物在Raney Ni陰極上的脫甲氧基化機理(圖8),發(fā)現(xiàn)愈創(chuàng)木酚脫甲氧基是由苯環(huán)局部加氫引發(fā)的,經(jīng)歷甲氧基消除反應后,生成苯酚。當甲氧基與羥基呈鄰位關系時,脫甲氧基化反應的選擇性可達99%以上;而當甲氧基與羥基呈對位關系時,脫甲氧基化反應的選擇性僅為50%左右。這表明羥基可能有助于脫甲氧基化反應。脫甲氧基化步驟也適用于具有多個甲氧基的芳香族化合物,如2,6-二甲氧基苯酚。2,6-二甲氧基苯酚轉化為愈創(chuàng)木酚的速度比愈創(chuàng)木酚轉化為苯酚的速度慢,說明芳香環(huán)的電子密度影響脫甲氧基速率,電子密度越大,脫甲氧基速率越小。
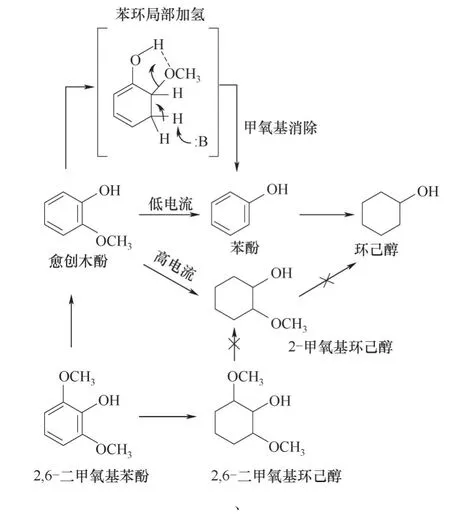
圖8 愈創(chuàng)木酚的電催化加氫反應機理Fig.8 Reaction mechanism of electrocatalytic hydrogenation of guaiacol
表3歸納了部分生物油模型化合物電催化加氫研究進展。通過上述研究,可總結出如下提升電催化加氫效率的途徑。降低催化劑顆粒尺寸可提高電導率,從而提高電化學效率。添加陽離子表面活性劑,可提高不飽和化合物的電催化加氫效率,其原因在于可在陰極形成附著在電極表面的疏水層,相對疏水的有機反應物在該層中的溶解度比H3O+更大,有利于提高有機物在催化劑表面的覆蓋率,強化加氫反應,減少析氫。降低生物油中的水分含量,有利于提高在催化點位有機反應物濃度,從而提高電化學效率。通過采用固體聚合物電解質體系,可減小電極間距并顯著降低溶液電阻,從而提升能源效率。
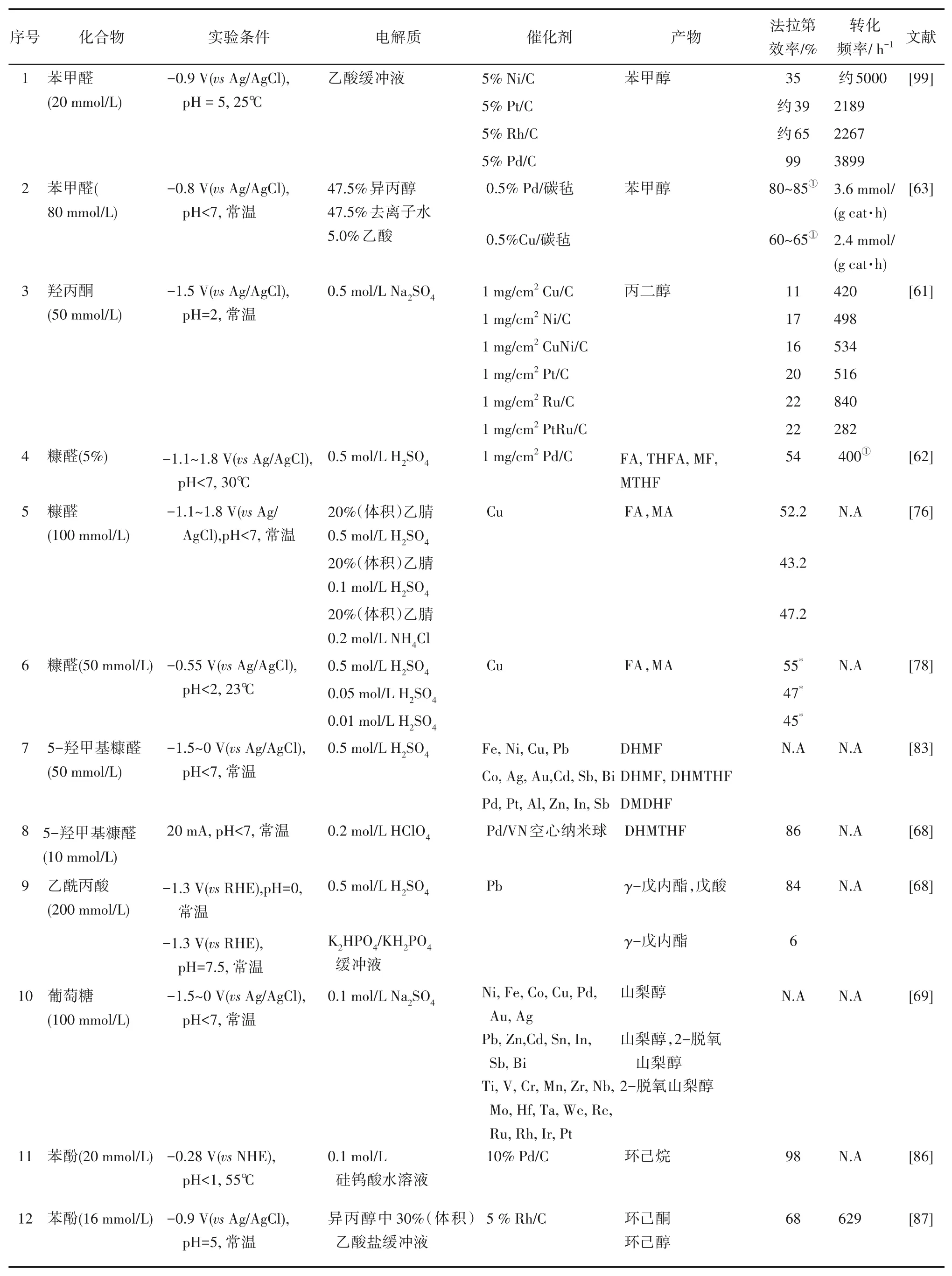
表3 生物油模型化合物電催化加氫研究Table 3 Studies on electrocatalytic hydrogenation of bio-oil derived model compounds
4 生物油電催化加氫
上述生物油模型化合物相關研究為進一步開展生物油電催化加氫研究提供了數(shù)據(jù)與理論支撐。與模型化合物研究相比,實際生物油樣品的電催化加氫研究仍然有限。一方面原因可能是由于缺乏熱解設備,相關研究團隊難以獲取質量穩(wěn)定的生物油用于研究。另一方面,生物油酸性高,含有大量具有反應活性的組分,其穩(wěn)定存儲仍然面臨挑戰(zhàn)。然而,仍有學者設法進行了生物油電催化加氫提質研究,并證明電催化加氫是提升生物油化學穩(wěn)定性的可行方法,相關研究歸納在表4中。實際生物油樣品的電催化加氫無法采用法拉第效率進行評估,這是由于生物油的化學組分復雜,很難在提質反應后的生物油中量化加氫產物,因而無法進行法拉第效率計算。因此,需要其他的方式來評估生物油的提質過程。例如,羰基濃度[59]和總酸值(TAN)[69]分別代表生物油中羰基和酸組分的含量,是常用的生物油性質評估參數(shù)。利用凝膠滲透色譜(GPC)測定生物油的分子量分布情況,是評價生物油提質前后穩(wěn)定性的有效方法之一。
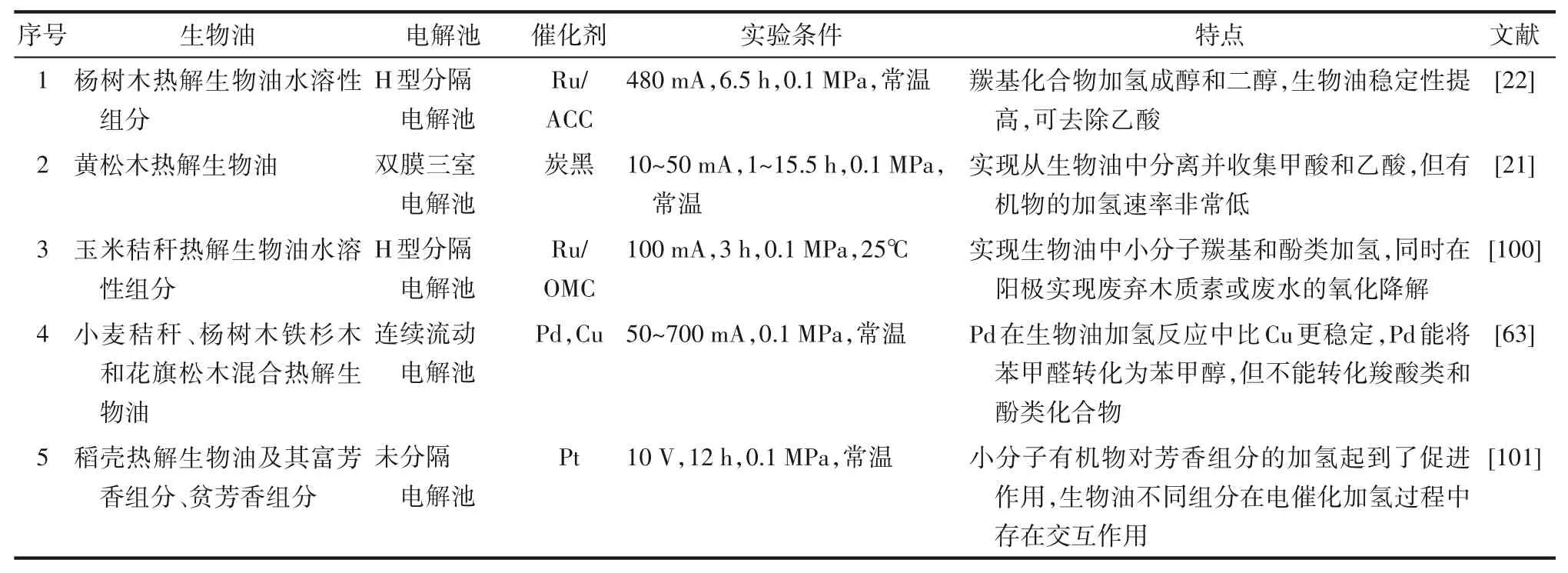
表4 生物油電催化加氫研究Table 4 Studies on electrocatalytic hydrogenation of bio-oil
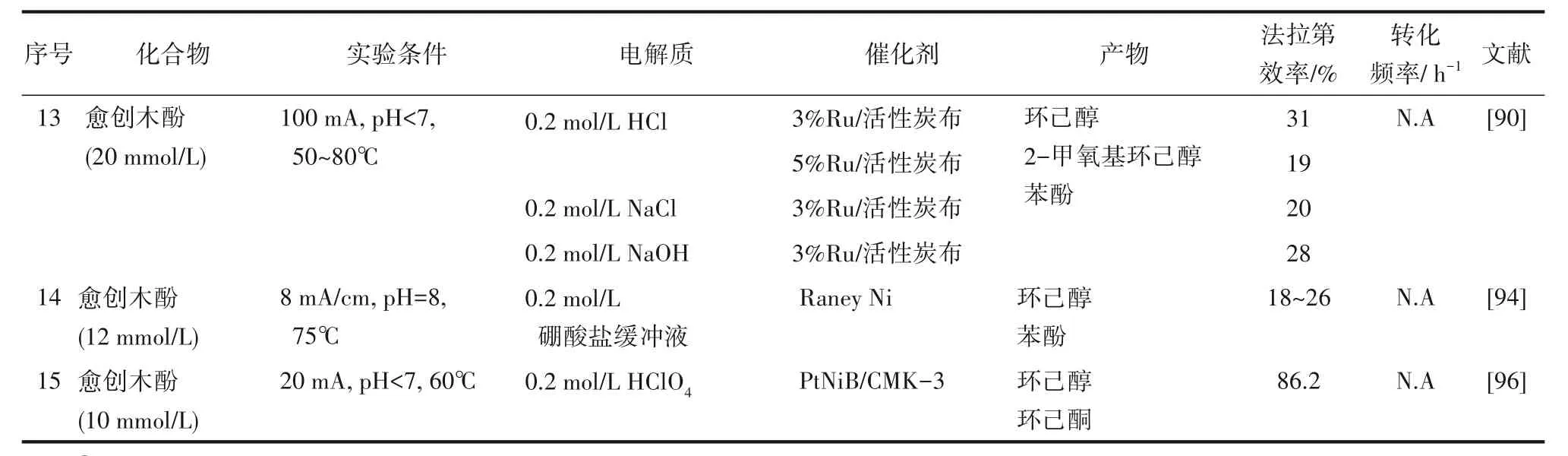
續(xù)表3
Li等[22]率先在活性炭布上負載Ru催化劑(Ru/ACC)對楊樹木熱解生物油的水溶性組分進行了電催化加氫研究,結果表明幾乎所有的小的羰基化合物均還原成相應的醇類和二醇類物質,并通過老化實驗證明了提質后的生物油穩(wěn)定性得到提高。研究采用H型電解池和質子交換膜。值得注意的是,提質后在陰極室中未檢測到乙酸,可能是帶負電的醋酸根離子由于陽極的吸引力而遷移到陽極室。
Lister等[21]在H型電解池基礎上,在質子交換膜和陽極之間增加一層陰離子交換膜,從而增加獨立中間腔室,設計了一種三腔室雙膜電解反應裝置。在電勢作用下,陽極側帶負電的酸根離子和陰極側的質子分別通過陰離子交換膜和質子交換膜進入中間腔室,成功將甲酸和乙酸分離并遷移進中間腔室,提高陰極室中生物油pH,并實現(xiàn)有機酸組分收集。然而,羰基濃度分析表明,生物油的加氫率較低,因此需要對電催化劑進行進一步研究,以提高該體系下的生物油加氫提質效率。
Zhang等[100]使用有序介孔碳負載釕催化劑(Ru/OMC)進行生物油水溶性組分電催化加氫研究。該體系能夠分別將羰基、酚類還原成醇、脂肪環(huán)。與Li等報道的Ru/ACC催化劑相比,Ru/OMC催化劑的釕顆粒尺寸更小,而且OMC的空腔可以保護釕金屬在實驗過程中不被溶解。另一個顯著特點是在陽極加入Fe3+和Fe2+氧化還原對作為氫載體,提高法拉第效率的同時,可利用廢水或木質素產生陰極生物油加氫所需的氫和電子,達到同時實現(xiàn)生物油加氫和降解廢水或木質素的目的。
Andrews等[63]進行了Cu和Pd金屬催化劑對生物油電催化加氫研究,目的是評價Cu和Pd在生物油電催化加氫中的性能和穩(wěn)定性。雖然Cu和Pd在苯甲醛加氫中表現(xiàn)出很高的效率,但對于生物油,只有Pd成功地降低了生物油樣品的羰基濃度,而Cu在生物油加氫反應過程中失活。這表明模型化合物研究可為揭示反應機理提供信息,但不足以預測實際生物油樣品的電催化加氫結果。
Deng等[101]將生物油全組分進行分離,分別對生物油全組分、富芳香組分和貧芳香組分進行了電催化加氫研究。實驗采用單室電解池,未采用離子交換膜。反應12 h后,觀察到貧芳香組分實現(xiàn)加氫,而富芳香組分更具有聚合傾向。當對生物油全組分進行處理時,芳香組分含量迅速下降,說明小分子有機物促進了芳香組分的加氫。這些小分子有機物可能作為有機共溶劑,加速芳香組分的加氫。當甲醇或乙醇被用作共溶劑時,在其他模型化合物的研究中可以觀察到類似的結果[102]。
5 結 語
生物油作為一種可再生的含碳原料在液體燃料生產中顯示出巨大的潛力。原始生物油必須經(jīng)過提質,降低其腐蝕性、提高其化學穩(wěn)定性,以便儲存或運輸。生物油電催化加氫技術是一種在溫和條件下提質生物油的新策略,近年來備受關注。電催化加氫可以避免生物油受熱結焦,在產物選擇性、工藝靈活性和效率方面具有顯著優(yōu)勢。目前,生物油電催化加氫提質技術處于實驗室探索階段,面臨一系列問題和挑戰(zhàn)。
與生物油模型化合物相比,實際生物油樣品的電催化加氫研究還處于初步探索階段,其原因主要有生物油固有的組分復雜性、生物油樣品的不一致性、生物油提質前后的表征困難以及產物定量難以實現(xiàn)。雖然模型化合物的研究有助于對反應機制和不同官能團之間的加氫位點競爭反應過程的理解,但真實生物油含有上百種化合物,在電催化加氫過程中構成復雜的反應網(wǎng)絡,且不同化合物之間的交互作用影響電催化加氫產物分布,僅憑單一的模型化合物難以客觀全面地反映真實生物油在電催化加氫過程中的演化機制。除了小分子化合物,生物油中還含有15%~30%(質量分數(shù))的大分子重質組分,這些組分在電催化過程中的演化機制不明晰。因此,加強對生物油全組分的電催化加氫提質研究顯得尤為重要。生物油中上百種化合物之間的交互作用以及它們在電催化加氫過程中對電極表面的競爭反應過程,導致反應監(jiān)測和分析極為困難。為此,諸如原位紅外光譜儀、原位拉曼光譜儀、電子自旋共振波譜儀(ESR)等原位測試技術有必要引入電催化研究中,配合定制的微小尺度連續(xù)取樣裝置,以測定電解液/催化劑/電極界面上的中間產物和自由基,從而獲取更多實驗信息。此外,生物油黏度高、電導率低,選擇合適的電解液(溶劑)和支持電解質是進行實驗研究的前提條件。需要注意的是,生物油起始樣品和最終產物都溶解在同一電解液中,從電解液中分離出目標生物油是一個重要環(huán)節(jié),相關分離技術值得探索。
通過量子化學模擬可以從分子層面深入剖析實驗難以解析的反應機理,目前關于生物油電催化加氫機制的量子化學模擬研究主要集中在小分子模型化合物,對于大分子模型化合物,由于其分子量大,存在模擬計算量大的問題,而生物油中的大分子重質組分由于結構未知,建模困難,難以對其電催化加氫機理進行仿真計算。精確解構重質組分大分子結構是進行仿真建模的前提,也是量子化學模擬研究生物油電催化加氫機制的重要方向。
生物油電催化加氫的反應條件是研究熱點之一。電解液酸堿度是影響電催化加氫性能的重要因素之一。有機分子在陰極上的吸附是電催化加氫反應的關鍵步驟,有機分子在不同酸堿環(huán)境下的陰極表面吸附能力不同,進而影響其加氫反應過程。酸性電解液中的中性分子更容易在陰極吸附,有利于電催化加氫反應;堿性電解液中具有離子狀態(tài)的有機物難以吸附,不利于電催化加氫反應。對于醛、呋喃、酚類,酸性條件有助于加氫反應[60,80,90]。若酸性過高,電解液中存在過量的H+可能會導致析氫副反應增強,也不利于加氫反應[85]。對于生物油,酸性環(huán)境下酚類、醛、呋喃以及大分子重質組分容易發(fā)生聚合,從而降低加氫效率。溫度是重要的影響因素,當溫度升高到一定程度,析氫反應活躍而降低了加氫效率。可見,電催化加氫的選擇性和效率的優(yōu)化依賴于電解液pH、電位、溫度和催化劑等多方面條件的協(xié)同作用。
電催化劑是提高產物選擇性和轉化率的關鍵。考慮到防止羰基化合物加氫過程中發(fā)生縮合,Pd和Ru電催化劑以其耐久性和優(yōu)越的加氫性能顯示出較強的應用前景。但由于儲備有限,其可持續(xù)性面臨挑戰(zhàn)。Cu對羰基官能團的加氫有很高的活性[63,68],但在實際生物油中不穩(wěn)定,因此,提高Cu的抗失活能力值得探究。研究表明采用金屬合金或設計配體可提高催化劑的耐久性,防止自腐蝕[103],例如Cu-Ni合金抗侵蝕性能優(yōu)異[104]。嘗試不同催化劑合成方法,如多孔納米結構催化劑基底、不同電極修飾方法等,也一直是電催化劑研究的熱點[22,99]。陽極鎳在電催化加氫反應過程中會氧化而被稱為犧牲陽極,從陽極腐蝕中浸出的鎳金屬離子會被重新沉積在陰極上,形成新的催化表面,因此鎳可提高呋喃或芳香族化合物電催化加氫的法拉第效率[80,105]。值得注意的是,Liu等[86]所提出的雙催化劑系統(tǒng)和Zhou等[96]制備的復合納米材料電催化劑都顯著提高了酚類化合物加氫產率,這為設計高效電催化劑的研究提供了新的思路。
質子交換膜的耐用性和成本是電催化加氫工藝實施的主要障礙之一。質子交換膜的作用是保護生物油中的易氧化成分不受陽極影響。由于生物油中有大量的小分子有機物和有機酸,膜容易受到有機污染而需要定期更換。經(jīng)濟分析研究表明,膜的成本回收期超過十年[106]。降低運行成本的研究方向之一是在陽極室中引入合適的有機基質(甲醇、5-羥甲基糠醛和甘油等),使其在提供氫源的同時氧化為高值化產品,從而提升整個電催化系統(tǒng)的經(jīng)濟性。
生物油電催化加氫具備顯著優(yōu)勢與工業(yè)化潛質,其能量來源形式廣泛(太陽能、風能等),工藝系統(tǒng)靈活,適合組成分布式系統(tǒng),同時也容易與現(xiàn)有的生物質熱解多聯(lián)產技術和石油煉油工藝整合。從能源角度看,生物質快速熱解制備生物油與電催化加氫提質策略耦合可以保留89%的生物能源[58]。因此,將生物質快速熱解和生物油電催化加氫相結合的生物質工業(yè)化利用技術路線極具前景。通過該技術路線,農林廢棄物可通過就地布置的熱解-電催化加氫耦合分布式系統(tǒng)[101]直接生產提質生物油,實現(xiàn)農林廢棄物等生物質資源的清潔利用,助力實現(xiàn)“2060碳中和”目標。
猜你喜歡
天天愛科學(2022年9期)2022-09-15 01:12:54
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天天愛科學(2022年4期)2022-05-23 12:41:48
當代水產(2022年3期)2022-04-26 14:26:56
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
航空世界(2020年10期)2020-01-19 14:36:20
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
