鈦硅分子篩TS-1催化環氧丙烷異構反應的機理探究
2021-10-31 23:36:46王剛段學志袁渭康周興貴
化工學報 2021年10期
關鍵詞:催化劑
王剛,段學志,袁渭康,周興貴
(華東理工大學化學工程聯合國家重點實驗室,上海 200237)
引 言
環氧丙烷(PO)是重要的有機化工原料,主要用于合成聚醚多元醇進而生產聚氨酯[1-2]。相比于工業上PO的生產方法,丙烯氫氧環氧化一步制備PO具有過程簡單、綠色環保、原子利用率高等優勢[3-7]。大量的含鈦材料被用于該反應,其中,鈦硅分子篩TS-1負載的納米金催化劑(Au/TS-1)因其優異的PO性能受到廣泛關注[8-12]。Zhou等[13-14]研究發現Au/TS-1催化劑快速失活的主要原因是結焦導致的微孔堵塞,提出了采用未焙燒的堵孔鈦硅分子篩負載納米金顆粒(Au/TS-1-B),結果表明可顯著提高催化劑的穩定性。值得指出的是,除了主產物PO外,反應過程中往往有丙醛等副產物的生成。通過對該催化劑上副產物的分布進行分析可發現,丙醛和丙酮的選擇性明顯高于其他副產物,且在反應過程中呈現逐漸上升的趨勢[15-16]。這些副產物的形成降低了PO的選擇性和催化劑長周期穩定性,為丙烯氫氧環氧化催化劑的設計與優化帶來了挑戰。Delgass等[17]對該反應進行了H2/D2同位素實驗,發現當原料氣由H2切換為D2后,除了丙烯醛外,各產物的生成速率均下降,并提出這些副產物可能是由PO發生深度反應轉化而來。Haruta等[18]通過動力學研究發現:隨著丙烯轉化率的增加,PO收率先上升后下降,而副產物的收率急劇上升,這預示著丙醛和丙酮可能是PO異構化的產物。Mul等[19]通過紅外光譜證實PO可轉化成雙配位丙氧基物種,并認為該中間體可進一步轉化為丙醛和丙酮副產物。對于PO在分子篩上的異構過程,Limtrakul等[20]采用理論計算研究了PO在ZSM-5分子篩酸性位上的異構機制,提出PO的異構主要存在兩個過渡態:碳氧環的質子化及斷裂;碳上氫原子的轉移重排。但目前對于PO在鈦硅分子篩上的異構機制尚未見報道,亟需對該過程進行機理性的認識與理解(圖1)。
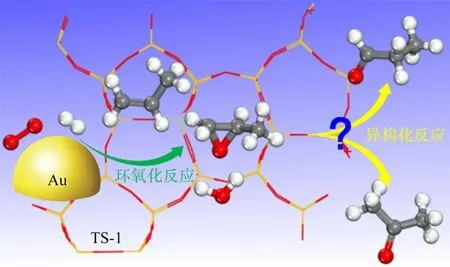
圖1 Au/TS-1催化丙烯氫氧環氧化生成PO以及PO異構化示意圖Fig.1 Schematic diagram of propylene epoxidation with H2 and O2 to PO and its isomerization over Au/TS-1 catalyst
本文首先制備了堵孔TS-1分子篩和Au/TS-1-B催化劑,并在固定床反應器中考察了其催化PO轉化的性能,進一步采用DFT理論計算研究了PO分子在TS-1的Ti-Defect位點附近轉化生成丙醛和丙酮的吸附態以及過渡態,提出了TS-1催化PO異構化的反應機理。這些研究結果將為丙烯氫氧環氧化催化劑上PO選擇性的進一步調控與優化提供理論依據。
1 實驗材料和方法
1.1 TS-1-B分子篩和Au/TS-1-B催化劑的制備
堵孔鈦硅分子篩的制備按照文獻中報道的水熱合成方法進行[9,13],具體步驟如下:在42℃下將2 g吐溫20溶于28 g超純水中,并加入質量分數為25%的四丙基氫氧化銨水溶液,攪拌過程中向溶液中加入40.5 g正硅酸乙酯。然后將0.67 g鈦酸四丁酯溶于15 g異丙醇溶液并添加到上述溶液中,利用水浴鍋將溶液升溫至80℃進行除醇。隨后將溶液轉移至水熱釜中并在170℃下晶化48 h。將產物進行離心和洗滌,并在100℃下干燥12 h得到白色固體,即為堵孔鈦硅分子篩,標記為TS-1-B。
Au/TS-1-B催化劑的制備參照文獻中報道的尿素沉積沉淀法(DPU)進行[17]。首先將1 g上述TS-1-B加入40 ml超純水中并攪拌,加入一定量的氯金酸的水溶液和尿素。隨后將該懸濁液升溫至90℃并保持6 h。隨后將固體和液體采用離心分離,用40 ml超純水洗滌,將獲得的固體在室溫下真空干燥得到未還原的金催化劑樣品。
1.2 樣品表征技術
采用X射線衍射(XRD)技術日本理學電機公司,18KW/D/MAX 2550 VB/PC對TS-1-B的物相結構進行分析,管電壓和管電流分別為40 kV和450 mA。
TS-1-B中Ti的配位狀態由紫外可見光譜(UVvis)美國Varian公司,Lambda 950型號紫外-可見-近紅外分光光度計進行分析。以硫酸鋇的光譜作為背景并扣除,儀器的波長范圍為175~3300 nm,UV-Vis波段的精度為±0.1 nm。
采用高分辨率透射電子顯微鏡(HRTEM),日本JEOL公司,JEM-2100型透射電子顯微鏡對分子篩的形貌和大小進行分析,其點和線分辨率分別為0.23 nm和0.14 nm。
采用電感耦合等離子原子發射光譜(ICPAES),美國安捷倫公司,Varian 710 ES型原子吸收光譜儀測定所合成的催化劑中Au和Ti的含量。測試之前先采用HF溶液和王水將催化劑溶解,然后進行測定分析,結合標準曲線和堵孔鈦硅分子篩中模板劑的含量約為16%[15]的結果,測得制備的Au/TS-1-B催化劑上Au元素的質量分數約為0.1%,分子篩中硅鈦比約為93。
催化劑上納米金顆粒的粒徑分布使用中國科學院蘇州納米所的高角環形暗場-掃描透射電子顯微鏡(HAADF-STEM)美國FEI公司Tecnai G2 F20 S-TWIN透射電子顯微鏡進行確定。
使用傅里葉變換紅外光譜(FT-IR)探究PO在TS-1-B分子篩和Au/TS-1-B催化劑上的吸附作用。使用儀器的漫反射模式,樣品池為Harrick公司生產的可通氣體的原位樣品池,使用液氮冷卻的Mercury-Cadmium-Telluride(MCT)檢測器記錄光譜信號。待樣品還原后降至室溫取背景,然后通入1.4%PO/He并保持30 min使其吸附飽和,隨后切換為Ar吹掃40 min并取光譜,二者作差譜獲得PODRIFTS結果。
1.3 性能考察
環氧丙烷異構化和深度氧化反應的性能考察在配有在線色譜儀的連續流動固定床裝置上進行。具體步驟如下:稱取0.15 g堵孔TS-1-B分子篩或Au/TS-1-B催化劑,將過篩后的樣品(<110μm)裝進內徑為6 mm的石英反應管中。采用40%的H2/N2混合氣對分子篩或催化劑預處理,流量為50 ml?min-1,升溫至300℃并保持2 h,調節反應器床層溫度至200℃并穩定一段時間后,調節氣體流量使PO體積分數為0.4%的PO/N2混合氣體通過床層進行反應,氫氣與氧氣的體積分數均為10%,總流量為35 ml?min-1,質量空速為14000 ml?h-1?g-1。采用在線氣相色譜儀分析反應器出口物質組成,其中PO、丙醛、丙酮和乙醛采用Restek公司生產的RT-QS-Bond色譜柱進行分離并通過氫離子火焰檢測器(FID)進行分析,H2和CO2采用TDX-01色譜柱進行分離并利用熱導檢測器(TCD)進行分析。各物質的相對校正因子均經過已知濃度的氣體進行標定。PO轉化率、產物選擇性以及碳平衡通過式(1)~式(6)進行計算。

式中,NPO,in為反應器進口中PO分子物質的量,mol;NPO,out、N丙醛,out、N丙酮,out、N乙醛,out、NCO2,out分別為反應器出口中PO、丙醛、丙酮、乙醛和二氧化碳分子物質的量,mol。
1.4 模型構建與計算方法
理論計算在Gaussian 09軟件包上進行,TS-1模型以MFI結構的SiO2分子篩為基礎截取團簇模型,將T6位點的Si原子替換為Ti原子,構造T5位點缺陷,團簇末端的Si懸斷鍵采用H原子進行飽和,并設置Si—H鍵的鍵長為0.146 nm[21-23]。計算中所使用的泛函和基組為B3lyp 6-31G(d,p),并結合D3BJ方法對體系的色散作用進行校正[24-26]。構建三足的Ti-Defect類型結構,即由一個鈦羥基與三個硅羥基組成,Wells等[21]研究發現該結構相比于四配位Ti(SiO)4位點具有更高的丙烯環氧化活性。計算中涉及的所有吸附態和過渡態均進行了頻率驗證,以確保吸附態沒有虛頻以及過渡態有且僅有一個虛頻,并對反應過渡態進行了IRC分析,驗證過渡態虛頻的振動方向為化學反應進行的方向。計算吸附能和過渡態的能壘采用各狀態的電子能量進行計算,并經過零點能和基組重疊誤差(BSSE)的校正。限制性優化后的9T分子篩模型結構如圖2所示,對該團簇模型中四個Ti—O鍵的鍵長進行分析(表1),發現它們介于0.175~0.182 nm之間,其平均值為0.179 nm,這一數值與文獻中EXAFS實驗所測得的(0.1793±0.0007)nm接近[27]。
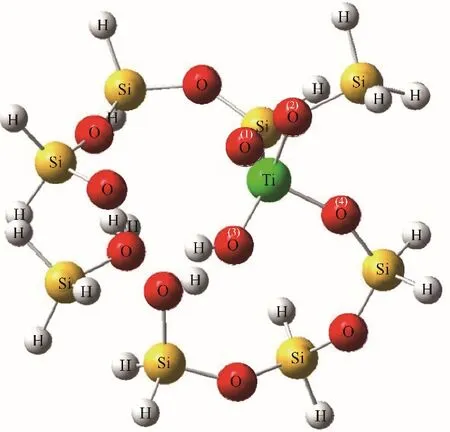
圖2 計算中所采用的TS-1分子篩9T模型Fig.2 The optimized 9T clusters model of TS-1 employed for the calculation

表1 TS-1模型中Ti—O鍵鍵長與實驗值比較Table 1 Comparison of Ti—O bond lengths between calculated and experimental values
2 實驗結果與討論
2.1 樣品表征與性能考察
圖3(a)TS-1-B的XRD譜圖表明其具有典型的MFI骨架結構[28-29]。采用UV-Vis光譜分析樣品中Ti的配位環境,圖3(b)中位于207 nm處的強吸收峰表明TS-1-B中鈦物種主要為四配位狀態[13],其中290 nm左右的肩峰歸屬于孔道內模板劑對鈦配位結構的影響[15]。圖3(c)HRTEM結果顯示所合成的分子篩大小較為均勻,粒徑為300~400 nm。采用氮氣物理吸附表征來探究所合成分子篩的比表面積,圖3(d)顯示了100℃干燥的堵孔TS-1-B和550℃焙燒的開孔TS-1-O的吸附等溫線,其中TS-1-O展現了典型的I型等溫線,表明其具有MFI的微孔結構。而TS-1-B由于微孔被堵塞而具有較少的氮氣吸附量。表2中給出了兩個樣品的BET比表面積和基于t-plot方法的孔容數據,結果顯示TS-1-B的BET比表面積約為39 m2?g-1,這與文獻中報道的TS-1-B的數據相一致[13]。
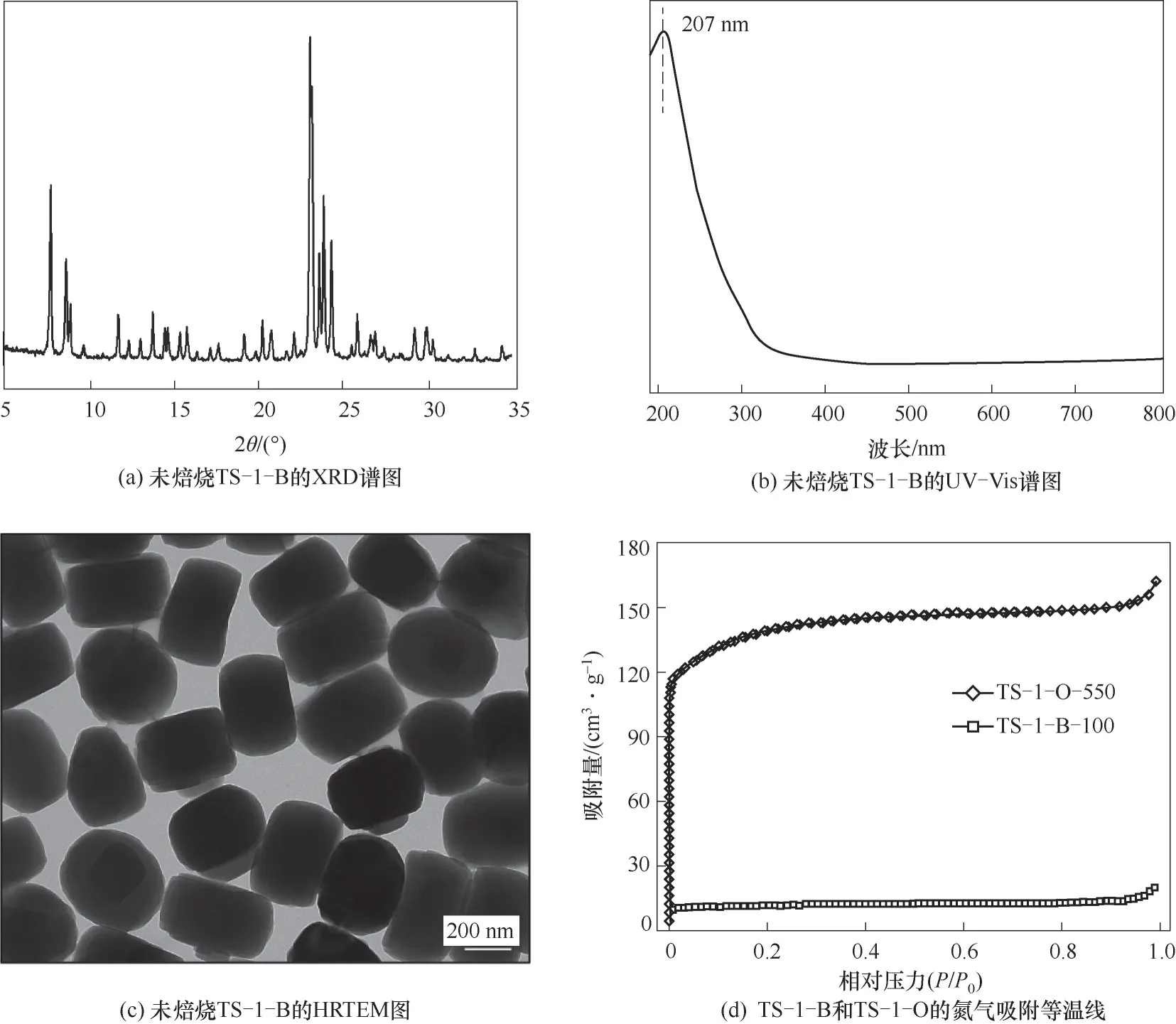
圖3 所合成的鈦硅分子篩的結構表征Fig.3 Structural characterization of synthesized titanosilicate

表2 TS-1-B和TS-1-O的BET比表面積和孔容參數Table 2 BET surface area and pore parameters of TS-1-B and TS-1-O
通過對還原后Au/TS-1-B催化劑的HAADFSTEM結果中超過200個金顆粒的粒徑統計,確定其粒徑分布約為(3.1±0.6)nm(圖4)。采用FT-IR探究PO在TS-1-B和Au/TS-1-B上的吸附作用,圖5(a)PO-DRIFTS結果顯示二者具有相似的PO吸附量,表明PO主要吸附在鈦硅分子篩而不是納米金顆粒上。采用不同氣氛下的反應結果來探究丙烯氫氧環氧化反應中丙醛和丙酮副產物的生成路徑。首先,向Au/TS-1-B催化劑上通入摩爾分數均為10%的丙烯與氧氣,發現在200℃的反應溫度下產物主要是丙烯醛,未檢測到明顯的PO、丙醛和丙酮。對該TS-1-B分子篩催化PO異構化的性能進行考察,結果如圖5(b)所示,觀察到產物中有丙醛和丙酮,且丙醛比丙酮具有更高的選擇性,這與該分子篩負載的納米金催化劑上的反應結果相吻合[15-16]。進一步向Au/TS-1-B催化劑上通入PO的結果如圖5(c)所示,可以看到催化劑上PO的轉化率略微升高,可能是由于金顆粒與載體之間的電子轉移作用促進了PO在分子篩上的吸附。而當通入氫氣和氧氣后,PO的轉化率急劇增加,并生成了乙醛和二氧化碳副產物,這可能主要是由于氫氣和氧氣在Au位點上反應生成的過氧物種與PO發生深度氧化反應,造成了碳碳鍵的斷裂生成乙醛和二氧化碳。這些結果說明了丙烯氫氧環氧化反應中丙醛和丙酮副產物主要來源于PO在分子篩上的異構化反應。
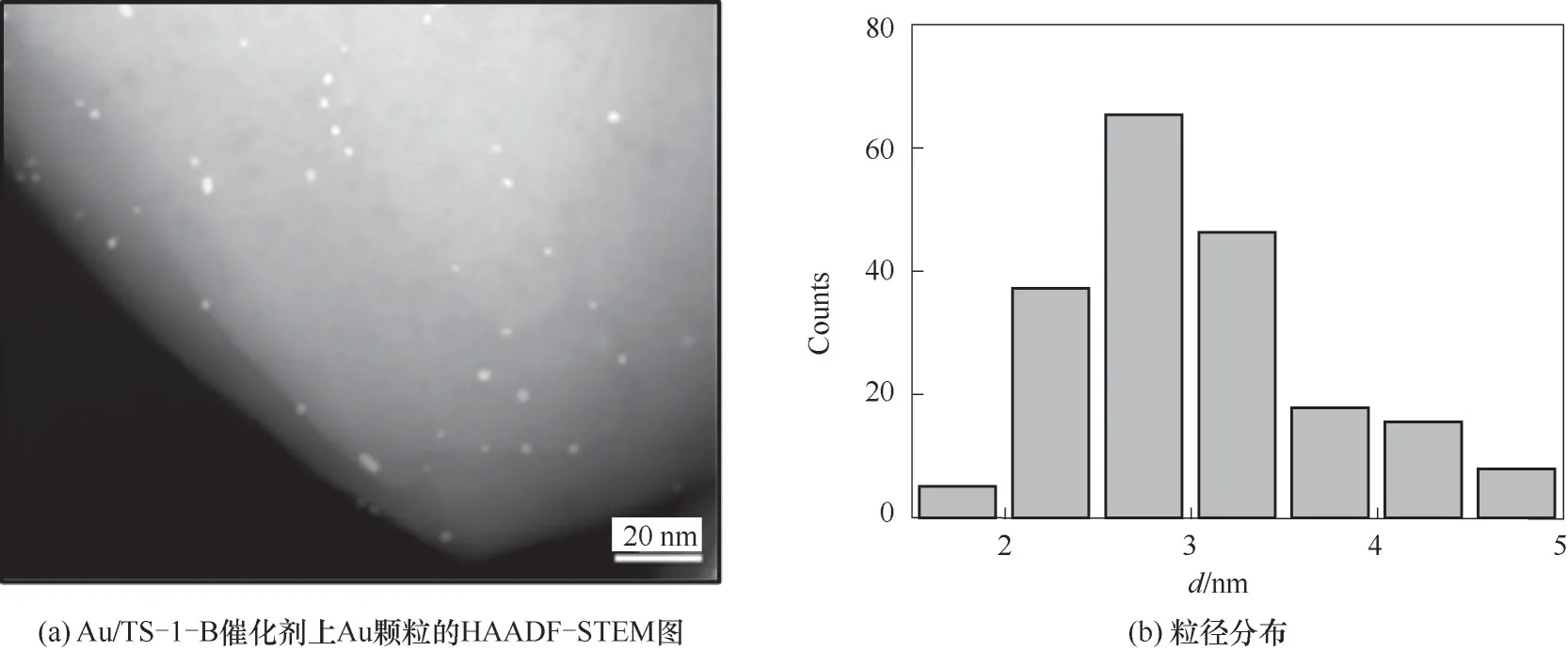
圖4 Au/TS-1-B催化劑上Au顆粒的HAADF-STEM和相應的粒徑分布Fig.4 Typical HAADF-STEM image and corresponding particle size distribution of Au/TS-1-B catalyst
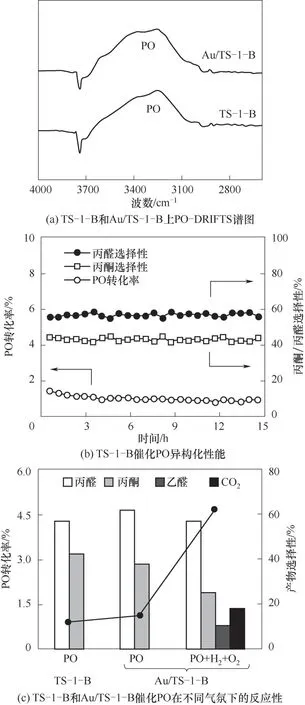
圖5 TS-1-B和Au/TS-1-B上PO異構化和深度氧化反應性能Fig.5 Catalytic performance of PO isomerization and overoxidation over TS-1-B and Au/TS-1-B
為了認識PO在TS-1分子篩上的開環和異構機理,采用理論計算探究PO分子在TS-1上轉化生成丙醛和丙酮的吸附態以及過渡態。通過對PO分子結構和反應路徑的解析可知,PO異構化的反應過程主要有碳氧鍵斷裂和氫原子轉移重排兩個過渡態[20,30]。由于PO分子中存在兩個C—O鍵,C1—O和C2—O鍵鍵長分別為0.143 nm和0.144 nm,斷裂后將分別朝向丙酮和丙醛的生成。下文將對這兩種情況分別進行計算研究和討論。
2.2 PO異構生成丙醛反應機理
PO異構生成丙醛過程中所涉及的關鍵結構和鍵長參數如圖6所示,PO首先吸附在TS-1酸性位點附近,可以看到PO分子主要通過其電負性的氧原子與Ti原子和羥基發生相互作用。通過分析吸附態PO中氧原子與其他原子的距離可知,PO與硅羥基上氫原子的距離(0.244 nm)比其與鈦羥基上氫原子的距離(0.279 nm)更近。這說明,相比于鈦羥基,PO與硅羥基具有更強的相互作用。動力學研究和紅外表征揭示這種吸附作用不僅占據了丙烯環氧化的活性位,對PO生成速率具有抑制效應,同時也會促進PO分子進一步開環異構和深度氧化生成副產物[31-33]。相比于氣相中C—O鍵鍵長,吸附態的鍵長得到了一定程度的拉伸,其中C2—O鍵的鍵長被拉伸至0.145 nm,預示著PO分子的活化,該吸附過程對于PO中三元環結構的碳氧鍵斷裂具有重要作用[20,34]。與二號碳原子相連的碳氧鍵在頻率振動中受到吸附作用的牽引,形成該化學鍵斷裂的過渡態,如圖6(b)所示,其中C2—O鍵的鍵長被進一步拉伸至0.215 nm。隨后與Ti相連的骨架中的氧原子牽引二號碳原子形成五元環的中間體結構,即雙配位丙氧基物種[圖6(c)]。大量光譜研究的結果證實了該物種在丙烯氫氧環氧化反應中的存在,其也被廣泛認為會進一步衍變成碳酸鹽或羧酸鹽物種并導致催化劑失活[19,35-38]。進一步,一號碳原子上的氫原子在頻率振動中受到二號碳原子的牽引,形成氫原子轉移重排的過渡態,同時C1—O鍵的鍵長縮短至0.132 nm,如圖6(d)所示。隨后二號碳原子捕獲到該轉移的氫原子,一號碳原子與氧原子形成鍵長為0.122 nm碳氧雙鍵,生成吸附態的產物丙醛。
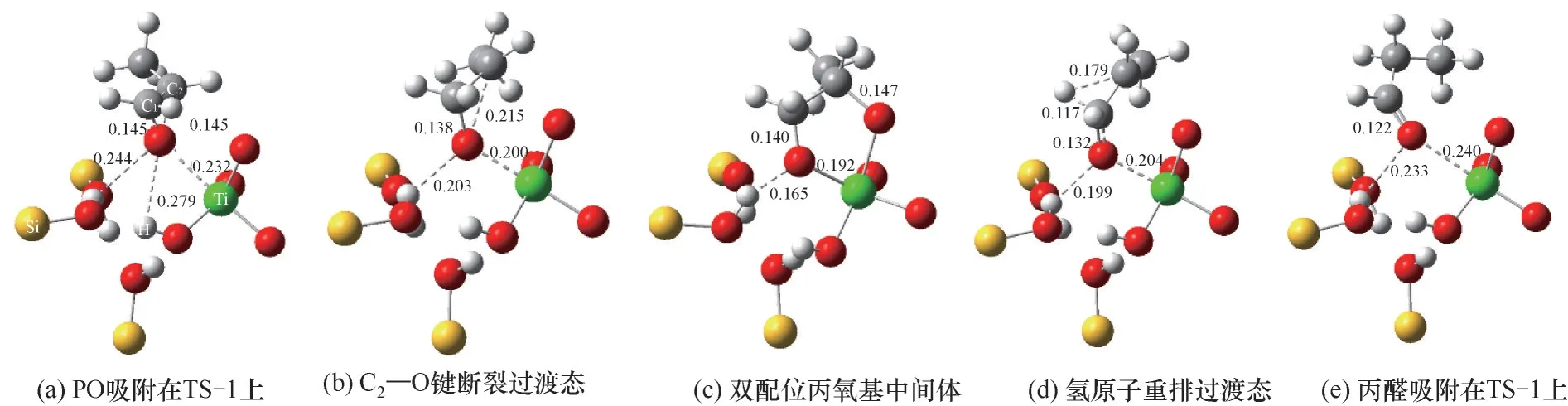
圖6 TS-1催化PO異構化生成丙醛中的關鍵結構和鍵長(單位:nm)Fig.6 The key structures and bond lengths during PO isomerization to produce propanal over TS-1(Unit:nm)
上述過程的能線圖如圖7所示,PO在該Ti位點上的吸附能為43 kJ?mol-1,C2—O鍵斷裂的過渡態與吸附態之間的能壘為152 kJ?mol-1,而碳上氫原子轉移重排的過渡態與吸附態之間的能壘為106 kJ?mol-1。通過對這兩個過渡態的頻率進行分析,發現其虛頻的振動頻率分別為-323 cm-1和-443 cm-1,從氣相反應物PO到氣相產物丙醛的過程中釋放能量約為96 kJ?mol-1。

圖7 TS-1催化PO異構化生成丙醛的能線圖Fig.7 Energy profile for PO isomerization to produce propanal over TS-1
2.3 PO異構生成丙酮反應機理
在PO異構形成丙酮過程中,PO在鈦位點附近與鈦原子、硅羥基以及鈦羥基發生吸附作用,并隨后經過碳氧鍵斷裂和氫原子轉移重排兩個過渡態。與PO異構形成丙醛所不同的是,丙酮的形成需要斷裂PO分子中一號碳原子上的碳氧鍵,并與鈦原子形成五元環結構的雙配位丙氧基物種中間體,隨后該物種二號碳原子上的氫原子發生重排向一號碳原子轉移,生成吸附態的產物丙酮,各關鍵步驟的結構和鍵長參數如圖8所示。

圖8 TS-1催化PO異構化生成丙酮中的關鍵結構和鍵長(單位:nm)Fig.8 The key structures and bond lengths during PO isomerization to produce acetone over TS-1(Unit:nm)
PO異構形成丙酮過程的能線圖如圖9所示,C1—O鍵斷裂和氫原子重排過程兩個過渡態的振動頻率分別為-454 cm-1和-435 cm-1,其與吸附態的能壘均比丙醛的生成過程對應的能壘要高,尤其是對于氫原子重排過程,達到了162 kJ?mol-1。這可能是由于丙酮生成過程中,二號碳原子上的氫向一號碳原子轉移時會受到三號碳原子反方向的吸引力,造成了過渡態的能量較高。從氣相反應物PO到氣相產物丙酮的過程中釋放能量約為130 kJ?mol-1。

圖9 TS-1催化PO異構化生成丙酮的能線圖Fig.9 Energy profile for PO isomerization to produce acetone over TS-1
通過把PO異構的兩條路徑整個過程進行對比可以看到,丙醛生成過程的能壘(152 kJ?mol-1)小于丙酮生成過程的能壘(162 kJ?mol-1),預示著丙醛相比于丙酮更容易生成。這一結果與前文中PO異構化結果以及文獻中Au/TS-1-B催化劑上丙醛的選擇性比丙酮高相一致[15-16],也與文獻報道中ZSM-5分子篩上PO異構化的理論計算結果相吻合[20]。
基于以上研究結果,PO在鈦硅分子篩表面與鈦原子和羥基具有較強的相互作用,這不僅會引起PO的開環反應轉化形成副產物,也可能會占據丙烯環氧化的活性位,從而抑制PO活性。目前已有相關的研究工作報道采用硅烷偶聯劑提高催化劑的疏水性來減弱PO吸附從而增強丙烯環氧化性能[39-41]。本文的研究結果將為鈦基催化劑的結構改性以增強PO脫附從而調控PO活性和選擇性提供理論依據。
3 結 論
本文基于TS-1-B分子篩和Au/TS-1-B催化劑上不同氣氛下的FT-IR和性能考察結果,發現丙烯氫氧環氧化反應中丙醛和丙酮的生成主要來源于PO在鈦硅分子篩上的異構化反應。進一步對TS-1催化PO異構反應的機理進行了研究,采用DFT計算方法優化了TS-1上Si缺陷的Ti-Defect位點結構,探究了PO異構生成丙醛和丙酮的反應路徑及其各狀態對應的能量。結果顯示PO異構化需經歷碳氧鍵斷裂、形成五元環結構雙配位丙氧基中間體和碳上氫原子轉移重排等關鍵步驟。其中,相比于丙醛,丙酮的生成過程中由于C2上氫原子向C1轉移重排的過渡態能壘較高導致了丙酮在PO異構化反應中更難生成。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
