ZrO2/SiO2催化劑的制備及其氧化脫硫性能研究
2021-11-26 01:36:38劉曉藝李秀萍趙榮祥張豪
化工學報 2021年11期
劉曉藝,李秀萍,趙榮祥,張豪
(遼寧石油化工大學石油化工學院,遼寧撫順113001)
引 言
針對環境污染問題,世界各國普遍制定了嚴格的環保法規來控制燃油的硫含量[1-2]。在歐美等發達國家,汽油和柴油的硫含量已控制在10 μg/g 以內。中國在國六標準中對硫含量的要求與這些發達國家一致。傳統加氫脫硫(HDS)工藝對脂肪族硫化物的脫硫效果較好,但對芳香族硫化物及其衍生物的脫除效果不理想[3]。此外,高溫、高壓以及使用昂貴的催化劑使得加氫工藝的成本急劇增加。近年來,生物脫硫(BDS)、吸附脫硫(ADS)、氧化脫硫(ODS)等非加氫脫硫技術開始引起人們關注[4-6]。特別是氧化脫硫技術,由于條件溫和、成本低、效率高,被認為是最有前途的脫硫技術之一[7]。
過渡金屬氧化物(WO3、MoO3、TiO2等)憑借高的氧化態和容易回收等優勢在油品氧化脫硫領域備受關注[8],但比表面積小、容易團聚等缺點限制了其工業化進程,因此,研究人員研發出了負載型催化劑。這種催化劑的載體通常具有良好的化學和熱穩定性以及較大的比表面積[9]。將過渡金屬氧化物負載到高比表面積的載體上,可以提升活性位點的暴露程度,改變催化劑的表面結構進而提高催化效率。例如Shen 等[10]以可再生的納米纖維素為模板合成了含鎢的氧化硅介孔材料。結果表明,該催化劑對于噻吩、二苯并噻吩和4,6-二甲基二苯并噻吩均具有較好的氧化脫硫活性。郝陽陽等[11]通過混合煅燒法制備了MoO3/MIL-101催化劑,并以乙腈為萃取劑、H2O2為氧化劑構建氧化脫硫系統,該體系能去除油品中99.0%的DBT。González 等[12]合成了MoO3/SBA-15催化劑,并以H2O2為氧化劑,在最優條件下對4,6-DMDBT 的脫除率高達99%。在眾多載體中,SiO2憑借其較高的比表面積與良好的吸附性能成為載體的首要選擇。鋯基材料在催化領域有著重要的應用,主要是因為鋯基材料具有適中的比表面積,獨特的酸堿雙功能特性,較強的氧化和還原性能以及高的熱穩定性等優點[13]。在氧化脫硫領域,鋯基材料主要作為氧化脫硫催化劑的載體[14-17],其催化性能體現在金屬有機框架材料方面[18-21]。最近,Zhang 等[22]合成了ZrP/MCM-41 催化劑,研究表明催化劑中的Zr4+可以將硫化物氧化成砜類。深入研究鋯基材料的氧化活性對于開發新型的氧化脫硫催化劑具有重要的意義。
低共熔溶劑(DES)是由一定化學計量比的季銨鹽和氫鍵供體組成的低共熔混合物,具有蒸氣壓低、不可燃、易于回收并且可循環使用等優點,廣泛應用于如納米材料、電化學、催化劑、氧化脫硫等領域[23-25]。近年來,在低共熔溶劑中合成納米材料正成為研究的熱點,特別是V2O5、ZnO2、TiO2和g-C3N4/Fe2O3等金屬氧化物[26-29]。相比于傳統溶劑,低共熔溶劑制備方法簡單且是綠色的,可以減少對環境的污染;另外可以對材料的形貌等進行調控以獲得優異的理化性能。
本文以己內酰胺-八水氧氯化鋯低共熔溶劑為添加組分,通過溶膠-凝膠法合成含鋯的硅膠,然后再經過高溫煅燒制備ZrO2/SiO2,并以ZrO2/SiO2為催化劑和吸附劑,H2O2為氧化劑,研究了模擬油中硫化物的脫除率。考察反應溫度、氧硫比與催化劑的加入量等因素的影響。在最佳條件下,對不同硫化物的脫除效果進行反應動力學分析,考察催化劑的循環使用性能并對反應機理進行探討。
1 實驗部分
1.1 試劑及儀器
正硅酸四乙酯(分析純, 天津市大茂化學試劑廠);濃鹽酸(分析純,國藥集團化學試劑有限公司);八水氧氯化鋯(分析純,阿拉丁試劑有限公司);H2O2(30%,遼寧泉瑞試劑有限公司);無水乙醇(分析純,國藥集團化學試劑有限公司);正辛烷(分析純,國藥集團化學試劑有限公司);二苯并噻吩(分析純,阿拉丁試劑有限公司);4,6-二甲基二苯并噻吩(分析純,阿拉丁試劑有限公司);苯并噻吩(分析純,阿拉丁試劑有限公司);己內酰胺(分析純,國藥集團化學試劑有限公司)。
X 射線衍射儀(D8 Advance,德國Bruker 公司);紅外光譜儀(NEXUS 870,美國Nicolet公司);XPS 能譜儀(K-Alpha,美國Thermo Fisher Scientific 公司);掃描電子顯微鏡(SEM,PhilipsXL 30,荷蘭Philips 公司);N2吸附-脫附(Micromeritics ASAP 2010 型自動吸附儀);微庫侖分析儀(WK-2D,江蘇江分儀器廠)。
1.2 催化劑的合成
按摩爾比6∶1稱取一定量的己內酰胺與八水氧氯化鋯,在80℃下加熱攪拌,直到完全熔化成黏稠液體,得到已內酰胺-八水氧氯化鋯低共熔溶劑。量取7 ml 無水乙醇和10 ml 正硅酸四乙酯攪拌后形成混合液。然后將0.202 g 低共熔溶劑超聲溶解在混合液中。再將稀釋后的鹽酸(4 ml 濃鹽酸和7 ml H2O)逐滴加入前者中,攪拌30 min,并放于水浴鍋中在60℃下放置4 h 進行溶膠-凝膠過程。得到的膠狀物在130℃下烘干5 h,研磨成粉狀,置于馬弗爐(通入氮氣保護)中550℃下煅燒3 h,得到2%-ZrO2/SiO2催化劑。在同樣步驟下,改變低共熔溶劑的加入量為0.416 g 和0.613 g 分別制得4%-ZrO2/SiO2和6%-ZrO2/SiO2催化劑,并在不加低共熔溶劑條件下獲得SiO2,負載型催化劑的合成過程見圖1。
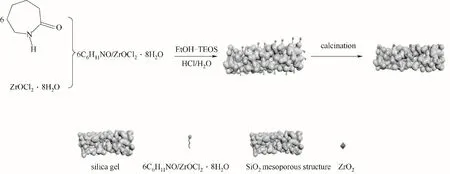
圖1 ZrO2/SiO2催化劑的合成過程Fig.1 The synthesis process of ZrO2/SiO2 catalyst
1.3 氧化脫硫過程
將1.437 g的DBT 加入500 ml的正辛烷中,配成含硫量為500 μg/g的模擬油,取5 ml模擬油,一定量的n-ZrO2/SiO2(n=2%,4%,6%) 和H2O2加入帶有冷凝裝置的磨口三角瓶中,在一定的溫度和設定轉速的磁力攪拌作用下進行氧化脫硫反應。每隔20 min取少量上層油相,通過微庫侖硫含量分析儀測定其硫含量,并用式(1)計算脫硫率。

式中,η為反應的脫硫率,%;C0與Ct分別為初始與t時模擬油中的硫含量。
2 結果與討論
2.1 催化劑的表征
2.1.1 FT-IR表征分析圖2顯示了ZrO2、SiO2、2%-ZrO2/SiO2、4%-ZrO2/SiO2和6%-ZrO2/SiO2的紅外譜圖。在SiO2的譜圖中,793 cm-1與1061 cm-1處吸收峰對應Si—O—Si 的對稱和非對稱伸縮振動,957 cm-1和1634 cm-1的吸收峰對應于Si-OH 的伸縮振動[30]。在ZrO2譜圖中,504 cm-1附近的吸收峰是Zr-O 鍵的伸縮振動峰[31],793 cm-1和452 cm-1處的吸收峰歸屬于Zr-O-Zr 鍵的彎曲振動[32]。3464 cm-1與1609 cm-1處的吸收峰是ZrO2吸附水的羥基振動造成的[33]。在ZrO2/SiO2的紅外譜圖中保持了氧化硅的特征峰,原ZrO2中504 cm-1和1609 cm-1處吸收峰消失,表明ZrO2在載體SiO2上高度分散。
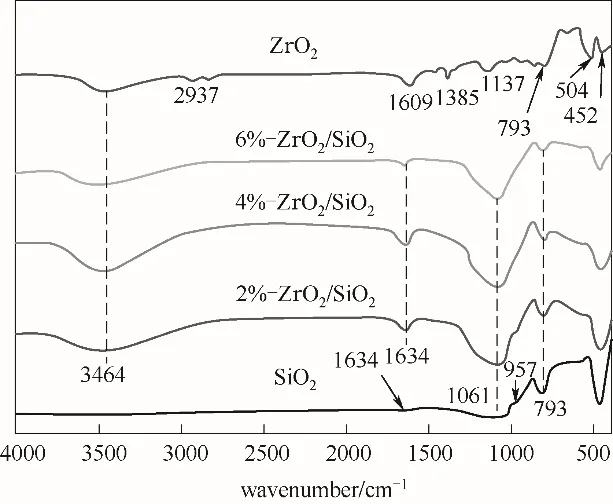
圖2 ZrO2、SiO2、2%-ZrO2/SiO2、4%-ZrO2/SiO2和6%-ZrO2/SiO2的FT-IR譜圖Fig.2 FT-IR spectra of ZrO2,SiO2,2%-ZrO2/SiO2,4%-ZrO2/SiO2 and 6%-ZrO2/SiO2
2.1.2 XRD 表征分析 圖3 顯示了SiO2與n-ZrO2/SiO2(n=2%,4%,6%) 的XRD 譜圖。圖中SiO2在2θ介于15°~35°之間存在一個較大的饅頭峰,其峰值為22.5°,說明合成的SiO2為無定形結構[22]。負載后的材料中未觀測到其他衍射峰,這可能是因為ZrO2在SiO2上分散度較高或者ZrO2含量太低。另外,在負載的幾種樣品中均觀測到一個較大的饅頭峰,這表明材料較好地保持了SiO2無定形結構。
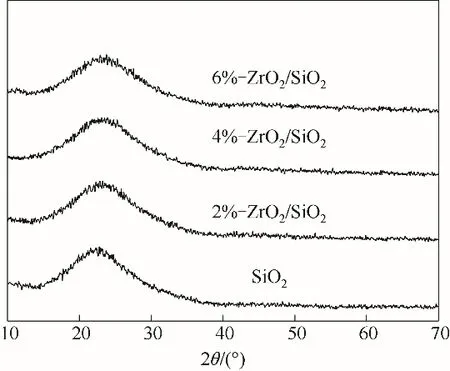
圖3 SiO2、2%-ZrO2/SiO2、4%-ZrO2/SiO2和6%-ZrO2/SiO2的XRD譜圖Fig.3 XRD patterns of SiO2,2%-ZrO2/SiO2,4%-ZrO2/SiO2 and 6%-ZrO2/SiO2
2.1.3 SEM 表征分析 圖4 為SiO2與4%-ZrO2/SiO2的掃描電鏡照片。從圖4(a)可以看出,正硅酸四乙酯在酸性條件下水解制備的SiO2呈現不規則的塊狀結構,表面比較光滑,未出現明顯的團聚現象。圖4(b)為負載后的樣品形貌,可以清晰看出在SiO2塊狀結構的表面聚集著大量的細小顆粒,表明了ZrO2已成功負載到SiO2上。

圖4 SiO2和4%-ZrO2/SiO2的SEM照片Fig.4 SEM images of SiO2and 4%-ZrO2/SiO2
2.1.4 BET表征分析 圖5(a)為SiO2、2%-ZrO2/SiO2、4%-ZrO2/SiO2和6%-ZrO2/SiO2的N2吸附-脫附曲線。根據曲線的趨勢判斷,幾種催化劑的吸附平衡等溫曲線均在0.39~0.65(P/P0)間出現了明顯的回滯環,屬于第Ⅳ類等溫吸附曲線,可知SiO2、2%-ZrO2/SiO2、4%-ZrO2/SiO2和6%-ZrO2/SiO2都屬介孔材料,此外,相對壓力較低時,氮氣吸附量有明顯上升趨勢,表明該材料也存在一定微孔。圖5(b)的孔徑分布曲線也證明載體和負載催化劑中存在微孔和介孔結構。
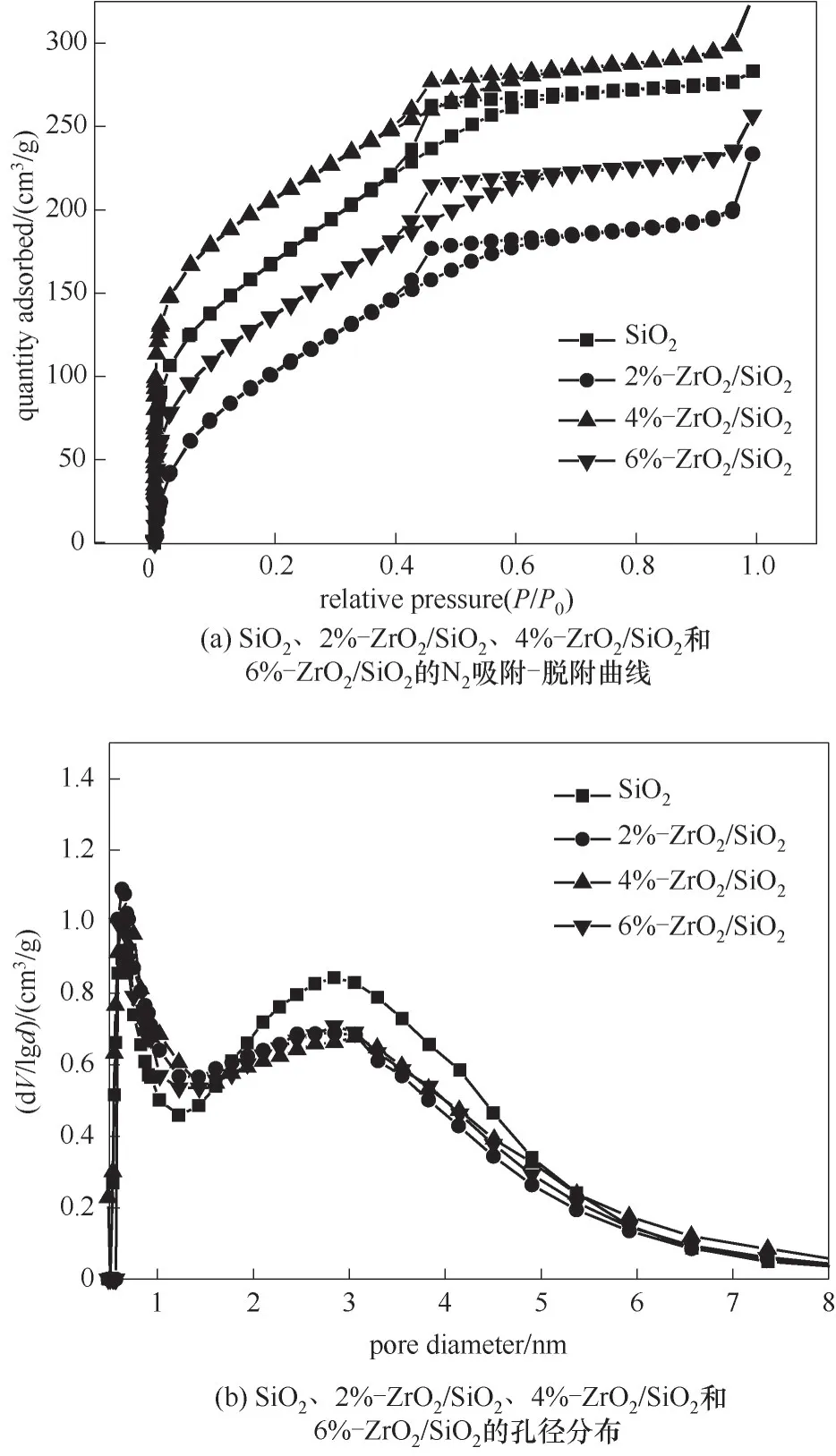
圖5 SiO2、2%-ZrO2/SiO2、4%-ZrO2/SiO2和6%-ZrO2/SiO2的N2吸附-脫附曲線和孔徑分布Fig.5 N2 adsorption-desorption isotherms and pore-size distribution curves of SiO2,2%-ZrO2/SiO2,4%-ZrO2/SiO2and 6%-ZrO2/SiO2
表1 為SiO2和n-ZrO2/SiO2(n=2%,4%,6%) 的比表面積及孔結構參數。如表所示,純SiO2的比表面積為618 m2/g,孔體積為0.7083 cm3/g。隨氧化鋯負載量的增加,除4%-ZrO2/SiO2外,其余樣品的比表面積出現不同程度的下降。一般認為,負載后催化劑表面積增加與低共熔溶劑的加入導致溶膠體系的表面張力改變有關[34]。負載后孔容均減小,可能是ZrO2顆粒進入SiO2孔道內填充造成的。純SiO2的孔徑為2.835 nm,負載后孔徑均有所增加,這可能是源于ZrO2填充了較小的孔隙,或者催化劑制備過程中高溫煅燒導致較薄的孔壁塌陷[35]。
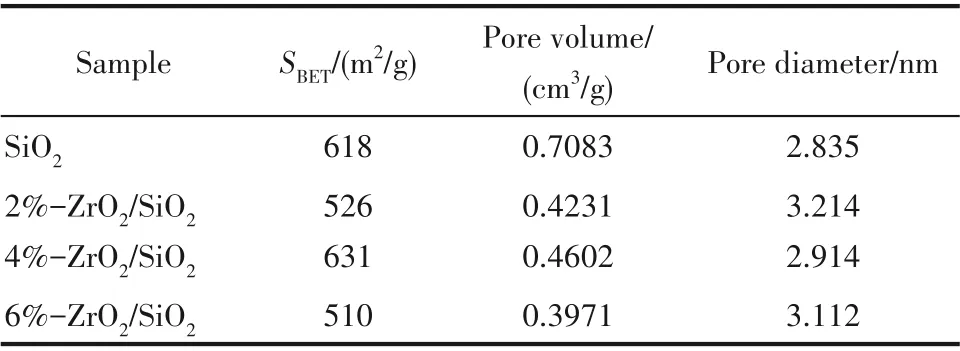
表1 樣品的比表面積及孔結構參數Table 1 Specific surface area and pore structure of samples
2.1.5 XPS表征分析 為了分析ZrO2/SiO2催化劑的元素組成與價態構成,對樣品進行了XPS 表征。圖6 為4%-ZrO2/SiO2的XPS 全譜圖與各元素的高分辨率XPS 譜圖。從圖6(a)可以看出,樣品中存在Si、O與Zr 三種元素,說明ZrO2已經負載到SiO2上。在Si元素的高分辨率譜圖中,103.4 eV 處有Si2p 的吸收峰,歸屬于Si4+,表明形成SiO2。與純SiO2中Si2p 的電子結合能103.7 eV 相比[24],其數值減小了0.3 eV,說明SiO2與ZrO2之間存在電子轉移。從圖6(c)中的氧元素的光電子能譜可知,樣品中的氧來源于Si—O(532.9 eV)、Zr—O(531.9 eV)和—OH(533.6 eV),與標準的SiO2與ZrO2的O1s 的結合能533.0 和530.4 eV[36-37]相比發生略微改變。在Zr 元素的高分辨XPS譜圖中,182.9 eV 與185.6 eV 處有兩個峰,分別與Zr3d5/2與Zr3d3/2相對應,其結合能與文獻報道相符[16,38],表明存在ZrO2。負載后ZrO2中Zr3d5/2的結合能182.9 eV 高于純ZrO2中Zr3d5/2的結合能182.5 eV,電勢向較大方向移動,可歸功于ZrO2分散在SiO2后兩者之間的相互作用[39]。因此,可以判斷,ZrO2被成功負載到了載體SiO2上。
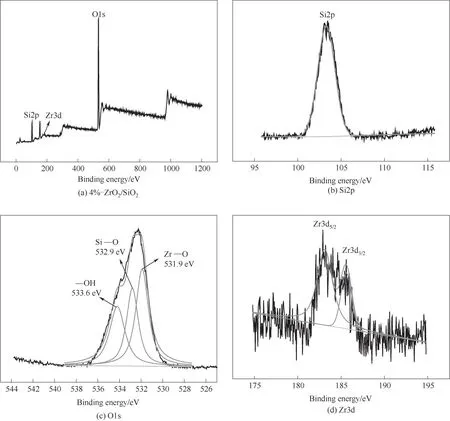
圖6 4%-ZrO2/SiO2的XPS譜圖Fig.6 XPS spectrum of 4%-ZrO2/SiO2
2.2 ZrO2負載量對脫硫效果的影響
在模擬油為5 ml,反應溫度為70℃,氧硫比為6的條件下,探究了氧化鋯負載量對DBT 脫除率的影響。如圖7 所示,當以純SiO2為催化劑時,DBT 的脫硫率為29%,這是因為煅燒后的SiO2吸附能力強,且具有一定的催化活性[40]。當氧化鋯的負載量為2%時,脫硫率為93.2%。隨著氧化鋯負載量增加到4%,其脫硫率上升到97.9%。繼續增加氧化鋯的負載量到6%時,DBT 的脫除率降至95.4%。以SiO2為催化劑時,盡管有較大的比表面積,但無活性組分ZrO2,所以脫硫率較低。相比于2%-ZrO2/SiO2,4%-ZrO2/SiO2比表面積從526 m2/g 增 加到631 m2/g,這意味著活性組分在載體上分散性更好,可以產生更多的活性位點。但是負載量繼續增加至6%時,過多的活性組分進入載體SiO2孔道中,導致比表面積下降并影響了對DBT 和氧化劑的吸附[41],導致脫除率下降。因此選擇4%-ZrO2/SiO2作為催化劑。
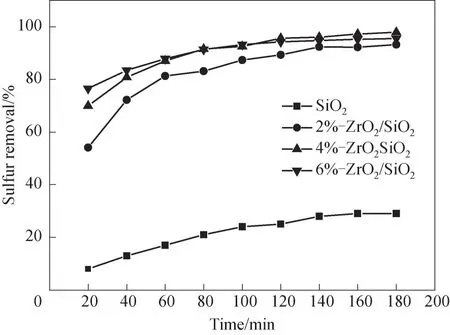
圖7 氧化鋯負載量對脫硫率的影響Fig.7 Influence of zirconia loading on desulfurization rate(reaction conditions:V(model oil)=5 ml,n(H2O2)/n(S)=6,T=70℃,m4% - ZrO2/SiO2=0.2 g)
2.3 反應溫度對脫硫效果的影響
反應溫度作為氧化脫硫反應條件中的重要因素之一,對脫硫率影響較大。如圖8所示,脫硫率以70℃為轉折點出現了先增加后降低的趨勢。當溫度為50℃與60℃時,DBT 的初始脫除速率較慢,180 min 后脫硫率分別達到95.8%和96.9%。當溫度提高到70℃時,反應速率明顯加快,脫硫率增長到97.9%。溫度升高到80℃時,雖然可以較快達到反應平衡,但DBT 的脫除率下降為93.5%。升高溫度可以使反應物分子間的碰撞加劇,加快反應進程[42];但過高的溫度也會導致雙氧水的分解速率加快,從而影響脫硫率[41-43],所以選擇70℃為最佳反應溫度。
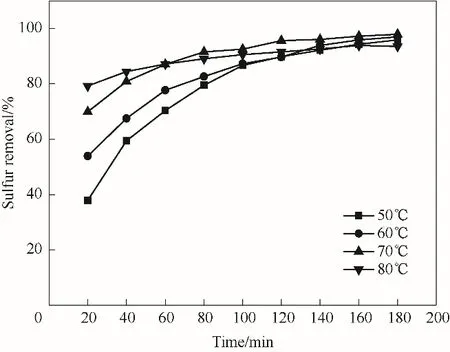
圖8 溫度對脫硫率的影響Fig.8 Influence of temperature on desulfurization rate(reaction conditions:V(model oil)=5 ml,n(H2O2)/n(S)=6,m4%- ZrO2/SiO2=0.2 g)
2.4 催化劑加入量對脫硫效果的影響
在氧化脫硫反應中,4%-ZrO2/SiO2作為吸附劑與催化劑,其加入量對脫硫率的影響較大。圖9 顯示了4%-ZrO2/SiO2加入量對脫硫率的影響。當加入量為0.1 g 時,反應180 min,脫硫率僅為45.2%。當4%-ZrO2/SiO2加入量為0.15 g 時,脫硫率提高到64.6%。當加入量增加到0.2 g 時,反應180 min 后DBT 的脫除率可達97.9%。 但繼續增加4%-ZrO2/SiO2量,DBT 的脫除率出現下降趨勢。一定范圍內,增加催化劑的加入量會使活性位的數量增多,導致更多的DBT 被氧化[44]。但當催化劑加入量過高時,載體上親水羥基官能團的數量也迅速增加,這些官能團大量捕獲H2O2,使得H2O2在活性位點附近與活性組分發生反應形成過氧化物的機會減少,導致脫硫率下降[35]。綜上,選擇4%-ZrO2/SiO2的加入量為0.2 g。
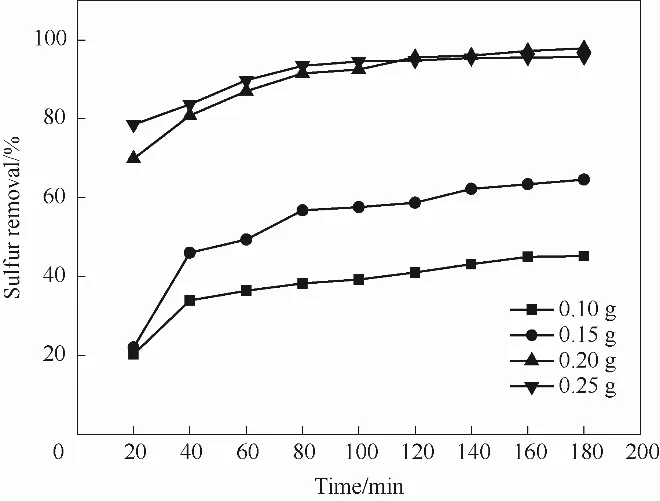
圖9 4%-ZrO2/SiO2的加入量對脫硫率的影響Fig.9 Influence of 4%-ZrO2/SiO2amount on desulfurization rate(reaction conditions:V(model oil)=5 ml,n(H2O2)/n(S)=6,T=70℃)
2.5 氧硫比對脫硫效果的影響
圖10顯示了雙氧水的量對脫硫率的影響,當氧硫比(摩爾比)為2 時,對DBT 的脫除率較低,僅為75.6%。當氧硫比增加到4 時,180 min 后DBT 的脫除率達到了98.7%,提升了23.2%。但氧硫比繼續增加到6 和8 時,脫硫率分別下降為97.9%與94.7%。這是因為來自雙氧水溶液中的H2O較多地吸附在催化劑的表面,從而導致活性位的減少以及有效比表面積的降低,從而抑制氧化脫硫反應的進程[35]。因此,選擇反應最佳氧硫比為4。
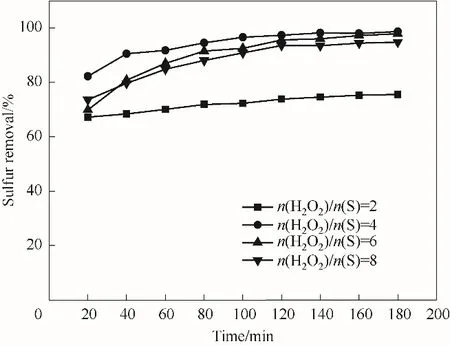
圖10 氧硫比對脫硫率的影響Fig.10 Influence of n(H2O2)/n(S)molar rate on desulfurization rate(reaction conditions:V(model oil)=5 ml,T=70℃,m4% - ZrO2/SiO2=0.2 g)
2.6 不同硫化物的脫除
由于真實柴油中含有BT、4,6-DMDBT、DBT 等多種硫化物,有必要考察催化劑對不同硫化物脫除的影響。考慮到催化劑本身對硫化物有吸附作用,考察了4%-ZrO2/SiO2分別對BT、4,6-DMDBT、DBT模擬油(均為500 μg/g)及真實柴油(硫含量稀釋為150 μg/g)的吸附脫硫效果,結果見圖11(a)。由圖可知,不同硫化物的吸附脫除效果為:DBT>4,6-DMDBT>BT>柴油。
圖11(b)顯示了最佳反應條件下4%-ZrO2/SiO2對BT 模擬油、4,6-DMDBT 模擬油、DBT 模擬油(硫含量均為500 μg/g)及含硫量為150 μg/g 柴油的氧化脫硫效果。結果顯示4,6-DMDBT 和DBT 脫除率較高,分別可以達到93.0%與98.7%,BT的脫除率較低,僅為65.9%。據報道[45],電子云密度較大的硫化物相對容易脫除。DBT、4,6-DMDBT、BT 相對應的電子云密度分別為5.758、5.760、5.739[46]。其中BT的電子云密度最小,所以最難脫除。而4,6-DMDBT盡管電子云密度較高,但是受空間位阻影響(含有兩個取代基)脫硫率小于DBT。該催化劑對柴油中硫化物的脫硫率較低(60.5%)是由于真實柴油成分復雜造成的。盡管如此,4%-ZrO2/SiO2催化劑仍可氧化脫除柴油中大部分硫化物,具有實際應用的潛力。
為了進一步了解氧化脫硫反應,評估催化劑對DBT、4,6-DMDBT 和BT 三種硫化物的脫除活性,有必要對脫硫過程進行動力學分析。先前的研究[47-48]表明氧化脫硫反應遵循一級反應動力學方程,因此用式(2)對不同硫化物的氧化脫硫反應進行動力學分析,其結果見圖12。

式中,k為一級反應動力學常數,min-1;t為反應的時間,min;C0為初始模擬油中硫化物的濃度,μg/g;Ct為時間t時模擬油中硫化物的濃度,μg/g。由圖12 可知,當用一級動力學方程模擬時,DBT、4, 6-DMDBT、BT 的動力學方程參數R2分別為0.95465、0.97614、0.98314,線性相關性良好。一級反應動力學常數k表示了該氧化脫硫反應進行的快慢,k值越大,反應速率越快,脫硫效果越好。DBT、4,6-DMDBT、BT 的k值分別對應為0.01574、0.00946、0.00365 min-1,即kDBT>k4,6-DMDBT>kBT。計算結果與圖11顯示的實驗結果一致。
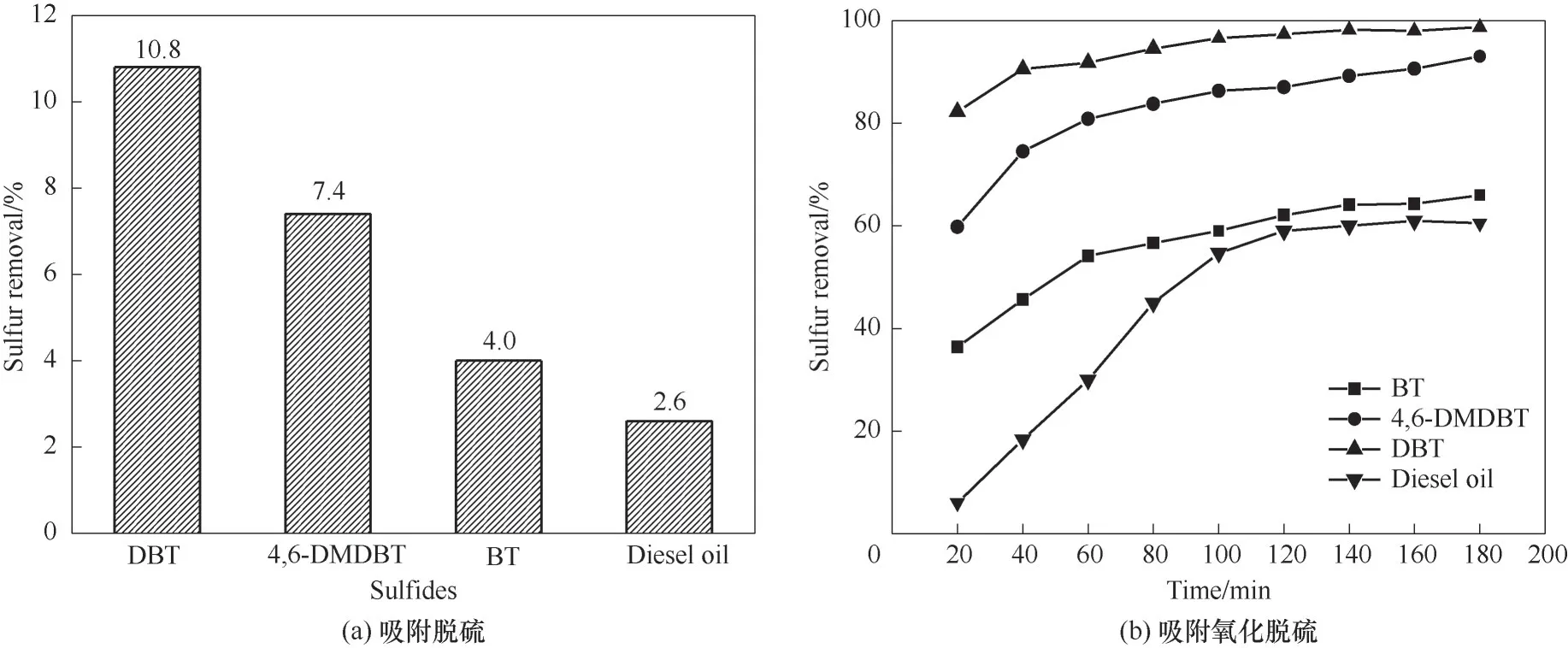
圖11 不同硫化物的脫除效果Fig.11 Removal efficiency of different sulfides(reaction conditions:(a)V(model oil)=5 ml,T=70℃,m4% - ZrO2/SiO2=0.2 g,t=180 min;(b)V(model oil)=5 ml,T=70℃,m4% - ZrO2/SiO2=0.2 g,n(H2O2)/n(S)=4)
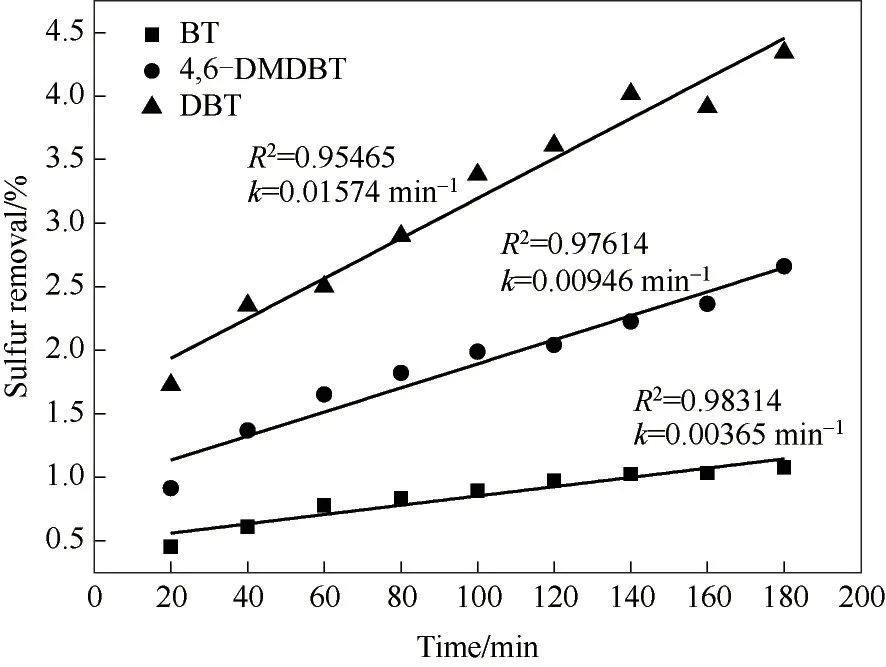
圖12 氧化脫硫反應中不同硫化物脫除的動力學分析Fig.12 Kinetic analysis of removal of different sulfides in oxidative desulphurization(reaction conditions:V(model oil)=5 ml,n(H2O2)/n(S)=4,T=70℃,m4% - ZrO2/SiO2=0.2 g)
2.7 ZrO2/SiO2催化劑的循環使用性能
為了考察催化劑的循環使用性能,在氧化脫硫實驗反應結束后,催化劑被離心分離并用無水乙醇和乙腈洗滌數次,并在110℃的烘箱中烘10 h,即可得到回收催化劑。在最佳反應條件下,加入新鮮雙氧水、模擬油以及回收的催化劑進行催化劑重復使用考察,實驗結果見圖13。結果表明,經過5次循環后,DBT 的脫除率下降為91.8%。這可能是活性組分的流失[10]及砜類物質在催化劑表面的堆積造成的[49]。
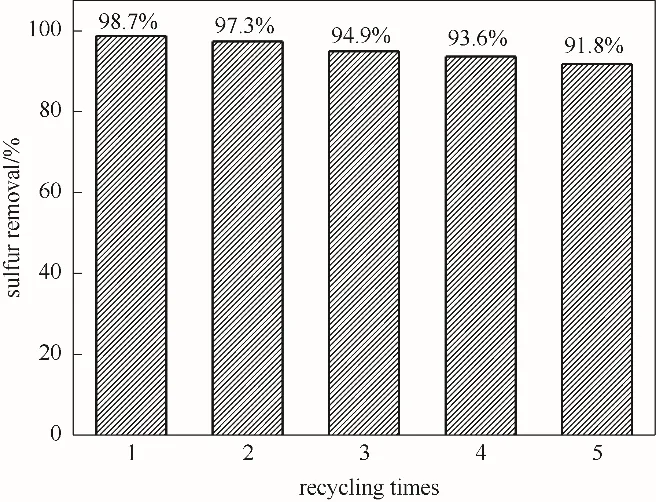
圖13 ZrO2/SiO2催化劑循環使用性能Fig.13 ZrO2/SiO2 recycling performance of catalyst(reaction conditions:V(model oil)=5 ml,n(H2O2)/n(S)=4,T=70℃,m4% - ZrO2/SiO2=0.2 g)
2.8 氧化脫硫機理
為了探討氧化脫硫反應機理,對使用前后的催化劑進行了紅外光譜分析并進行了自由基捕捉實驗。如圖14所示,使用后的催化劑上出現了二苯并噻吩砜的特征吸收峰,表明二苯并噻吩氧化成了砜并吸附到催化劑上。在氧化脫硫體系中分別加入0.0084 g 對苯醌(超氧自由基捕獲劑)和6 μl 異丙醇(羥基自由基捕獲劑)對DBT 模擬油進行氧化脫硫反應,實驗結果見圖15。由圖可看出,加入異丙醇后的脫硫率為88.6%,而加入對苯醌后的脫硫率僅為35.6%,可以判定,4%-ZrO2/SiO2氧化脫硫體系生成了超氧自由基·O-2和·OH, 并且·O-2在氧化脫硫中起到了關鍵作用,這與文獻報道一致[19,50]。圖16為ZrO2/SiO2脫硫機理,載體SiO2較大的比表面積使ZrO2的分散更均勻,也增加了DBT 與氧化劑的接觸概率。而ZrO2具有Lewis 酸性,DBT 中的硫原子含有孤對電子,可以作為電子供體或者Lewis 堿,二者之間存在相互作用,有利于油相中的DBT 吸附到催化劑上[51]。H2O2在ZrO2的作用下可以產生氧化能力較強的·和·OH 自 由基[52],從而將DBT 氧化成DBTO2并從油品中脫除。
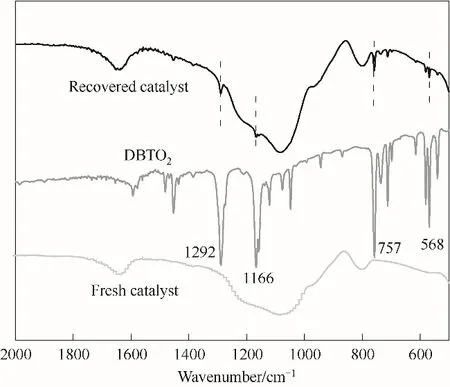
圖14 新鮮催化劑與回收催化劑的紅外譜圖Fig.14 Infrared spectra of fresh and recovered catalysts(reaction conditions:V(model oil)=5 ml,n(H2O2)/n(S)=4,T=70℃,m4% - ZrO2/SiO2=0.2 g)
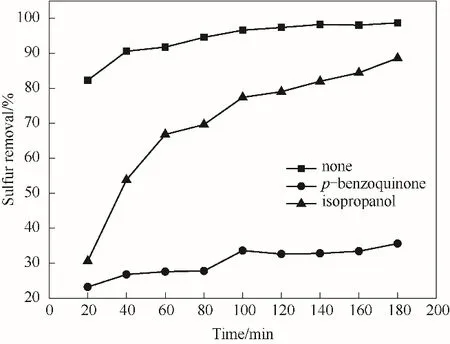
圖15 自由基捕捉實驗Fig.15 Free radical capture experiment(reaction conditions:V(model oil)=5 ml,n(H2O2)/n(S)=4,T=70℃,m4% - ZrO2/SiO2=0.2 g)

圖16 ZrO2/SiO2脫硫機理Fig.16 Desulfurization mechanism of ZrO2/SiO2
3 結 論
以己內酰胺-八水氧氯化鋯低共熔溶劑為添加組分,通過溶膠-凝膠過程合成了含鋯的硅膠,然后通過高溫煅燒制備了ZrO2/SiO2。采用XRD、FT-IR、XPS 等技術對其進行表征分析,結果表明成功制備了ZrO2/SiO2復合材料。并以H2O2為氧化劑、n-ZrO2/SiO2為吸附劑與催化劑對模擬油中的硫化物進行脫除研究。實驗結果表明,在模擬油為5 ml,溫度為70℃,n(H2O2)/n(S)=4,4%-ZrO2/SiO2量為0.2 g 時,對DBT、4,6-DMDBT 和BT 的脫除率分別為98.7%、93%和65.9%。催化劑經5 次再生使用后DBT 的脫除率仍可達到91.8%。在催化劑作用下雙氧水產生超氧自由基是硫化物被氧化的關鍵因素。
猜你喜歡
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
應用化工(2014年3期)2014-08-16 13:23:50
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
