訶子總黃酮提取工藝的優化及其體外生物活性研究
2021-11-26 01:31:50李國峰陳海芳郎一帆樊慧芳陳春蘭張武崗
中成藥 2021年11期
關鍵詞:黃酮
李國峰, 陳海芳, 郎一帆, 樊慧芳, 陳春蘭, 張武崗*, 付 丹
(1.江西中醫藥大學中藥固體制劑制造技術國家工程研究中心,江西 南昌 330006;2.江西中醫藥大學現代中藥制劑教育部重點實驗室,江西 南昌 330004;3.南昌大學第一附屬醫院,江西 南昌 330000)
黃嘌呤氧化酶(XOD)是嘌呤代謝過程中的關鍵酶,能催化黃嘌呤和次黃嘌呤氧化生成尿酸并產生自由基[1]。當體內尿酸濃度過高時,可導致高尿酸血癥并引起痛風發作,同時體內自由基的積累會導致脂質、DNA、蛋白質等生物大分子損傷,從而誘發疾病的發生發展,如糖尿病[2]、慢性腎病[3]、心血管疾病[4]等。因此,從中藥中尋找有效調節(抑制)XOD活性的成分是當前研究熱點[5]。
訶子為使君子科植物訶子TerminaliachebulaRetz.、絨毛訶子TerminaliachebulaRetz.var.tomentellaKurt.的干燥成熟果實[6],可用于治療久瀉久痢、便血脫肛、肺虛喘咳、咽痛音啞等疾病,被稱為“藥物之王”,主要含有鞣質類、多酚類、多糖類、揮發油類等[7-9]化合物,其提取物具有保肝[10]、降糖[11]、抗腫瘤[12]、抗炎[13]、解毒[14]等作用,但目前對該藥材藥理活性的研究僅限于總提物,尚未涉及其總黃酮對XOD的抑制作用和抗氧化活性。因此,本實驗采用響應面法優化訶子總黃酮制備工藝后,考察該成分對XOD的抑制作用和自由基的清除能力,以期為今后它在治療高尿酸血癥、痛風中的應用研究提供依據。
1 材料
1.1 試劑與藥物 訶子購自四川中庸藥業有限公司,經江西中醫藥大學楊世林教授鑒定為使君子科植物訶子TerminaliachebulaRetz.的干燥成熟果實。蘆丁對照品(成都曼思特生物科技有限公司,批號MUST-16031610);2,2-聯苯基-1-苦基肼基(批號C10544590)、維生素C(批號C10250787)、對氨基苯磺酸(批號C10540076)、鹽酸萘乙二胺(批號C10636097)、黃嘌呤(批號C10314543)、1,10菲羅啉(批號C10451583)、無水硫酸亞鐵(批號C10632300)(上海麥克林生化科技有限公司);黃嘌呤氧化酶(美國Sigma公司,批號SLCB1290)。亞硝酸鈉、硝酸鋁、氫氧化鈉、30%H2O2、無水乙醇等均為分析純;蒸餾水、超純水均由江西中醫藥大學現代中藥制劑教育部重點實驗室提供。
1.2 儀器 GZX-9140 MBE數顯鼓風干燥箱(上海博迅實業有限公司醫療設備廠);JA2003N電子天平(上海佑科儀器儀表有限公司);電子分析天平[賽多利斯科學儀器(北京)有限公司];L-500低速離心機(湖南湘儀實驗室儀器開發有限公司);HH-S恒溫水浴鍋(江蘇省金壇市醫療儀器廠);116搖擺式六兩裝高速中藥粉碎機(浙江上虞市道墟寶民儀器設備廠);酶標儀(美國Thermo公司);BUCHI Rotavapor R-200旋轉蒸發器(廈門精藝興業科技有限公司)。
2 方法
2.1 線性關系考察 參考文獻[15]報道的方法,略有改動。精密稱取蘆丁對照品25.0 mg,置于50 mL棕色量瓶中,60%乙醇溶解定容,即得500 μg/mL對照品溶液,置于4 ℃冰箱中保存備用。分別吸取0、0.2、0.4、0.6、0.8、1.0、1.2 mL至10 mL具塞試管中,加入5%NaNO2溶液0.3 mL,搖勻,靜置6 min,加入10%Al(NO3)3溶液0.3 mL,搖勻,靜置6 min,加入4%NaOH溶液5 mL,混勻,60%乙醇定容后靜置20 min,于510 nm波長處測定吸光度。以蘆丁質量濃度為橫坐標(X),吸光度為縱坐標(A)進行回歸,得方程為A=0.008 9X-0.013 9(R2=0.999 0),在0~60 μg/mL范圍內線性關系良好。

2.3 方法學考察
2.3.1 精密度試驗 精密吸取500 μg/mL對照品溶液0.6 mL,按“2.1”項下方法測定吸光度6次,計算得RSD為0.14%,表明儀器精密度良好。
2.3.2 穩定性試驗 精密稱取脫脂后的干燥藥材粉末5.0 g,按“2.2”項下方法提取總黃酮后,于0、1、2、4、8 h按“2.1”項下方法測定吸光度,計算得RSD為1.35%,表明提取液在8 h內穩定性良好。
2.3.3 重復性試驗 精密稱取脫脂后的干燥藥材粉末5.0 g,按“2.2”項下方法平行制備6份提取液,按“2.1”項下方法測定吸光度,計算得RSD為1.78%,表明該方法重復性良好。
2.3.4 加樣回收率試驗 精密稱取脫脂干燥后的總黃酮含量已知的藥材粉末6份,按80%水平加入對照品溶液,按“2.2”項下方法制備6份提取液,按“2.1”項下方法測定吸光度,計算回收率,計算得RSD為1.67%。
2.4 單因素試驗 精密稱取脫脂后的干燥藥材粉末5.0 g,置于150 mL具塞錐形瓶中,分別考察乙醇體積分數40%、50%、60%、70%、80%,液料比5∶1、10∶1、15∶1、20∶1、25∶1,提取時間30、60、90、120、150 min,提取溫度50、60、70、80、90 ℃對總黃酮提取率的影響。
2.5 響應面法 以總黃酮提取率(Y)為評價指標,乙醇體積分數(A)、液料比(B)、提取溫度(C)、提取時間(D)為影響因素,設計4因素3水平響應面實驗,平行3次,因素水平見表1。
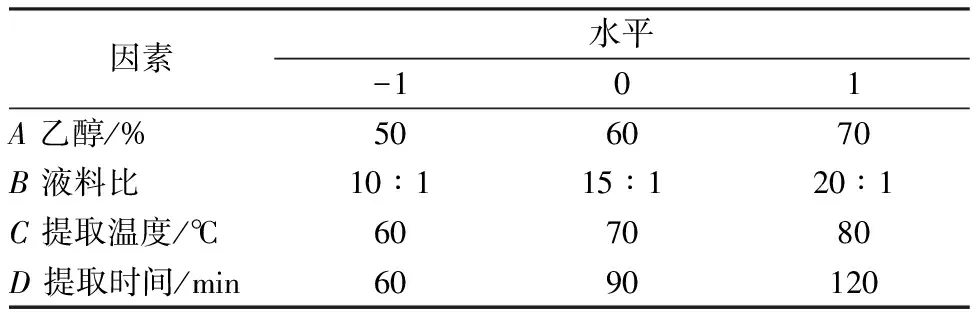
表1 因素水平
2.6 體外生物活性研究


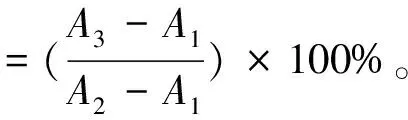
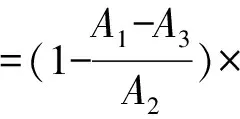
3 結果
3.1 單因素試驗
3.1.1 乙醇體積分數 如圖1所示,總黃酮提取率隨著乙醇體積分數增加呈先升后降的趨勢,在60%時最高,之后出現明顯下降趨勢。因此,確定乙醇體積分數為60%。
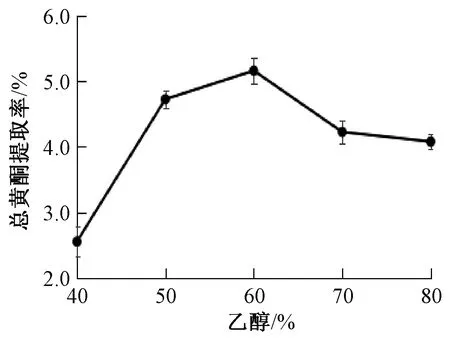
圖1 乙醇體積分數對總黃酮提取率的影響
3.1.2 液料比 如圖2所示,隨著液料比增加總黃酮提取率升高,在15∶1時最高,而大于15∶1后趨于平緩。因此,從節約成本方面考慮,確定液料比為15∶1。
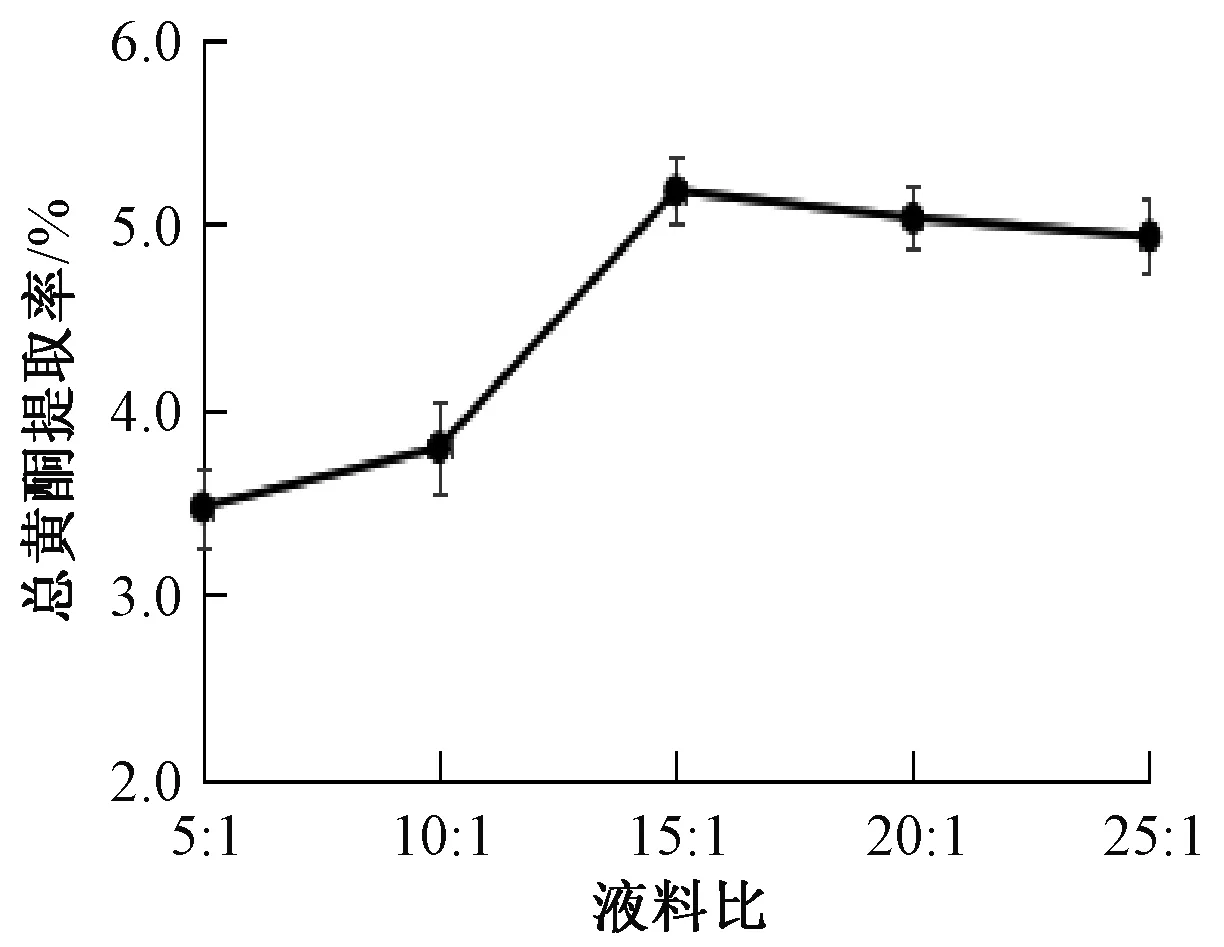
圖2 液料比對總黃酮提取率的影響
3.1.3 提取溫度 如圖3所示,隨著提取溫度增加總黃酮提取率顯著升高,在70 ℃時最高,而超過70 ℃后呈現下降趨勢,其原因可能為溫度過高會使對溫度敏感的成分降解為小分子,同時總黃酮結構中的酚羥基會發生氧化反應。因此,從能源、提取率方面考慮,確定提取溫度為70 ℃。

圖3 提取溫度對總黃酮提取率的影響
3.1.4 提取時間 如圖4所示,隨著提取時間延長總黃酮提取率逐漸升高,在90 min時最高,而超過90 min后趨勢不明顯,其原因可能為提取時間越長,組織細胞內外濃度差越小,同時部分產物也會發生分解。因此,從能源、效率方面考慮,確定提取時間為90 min。
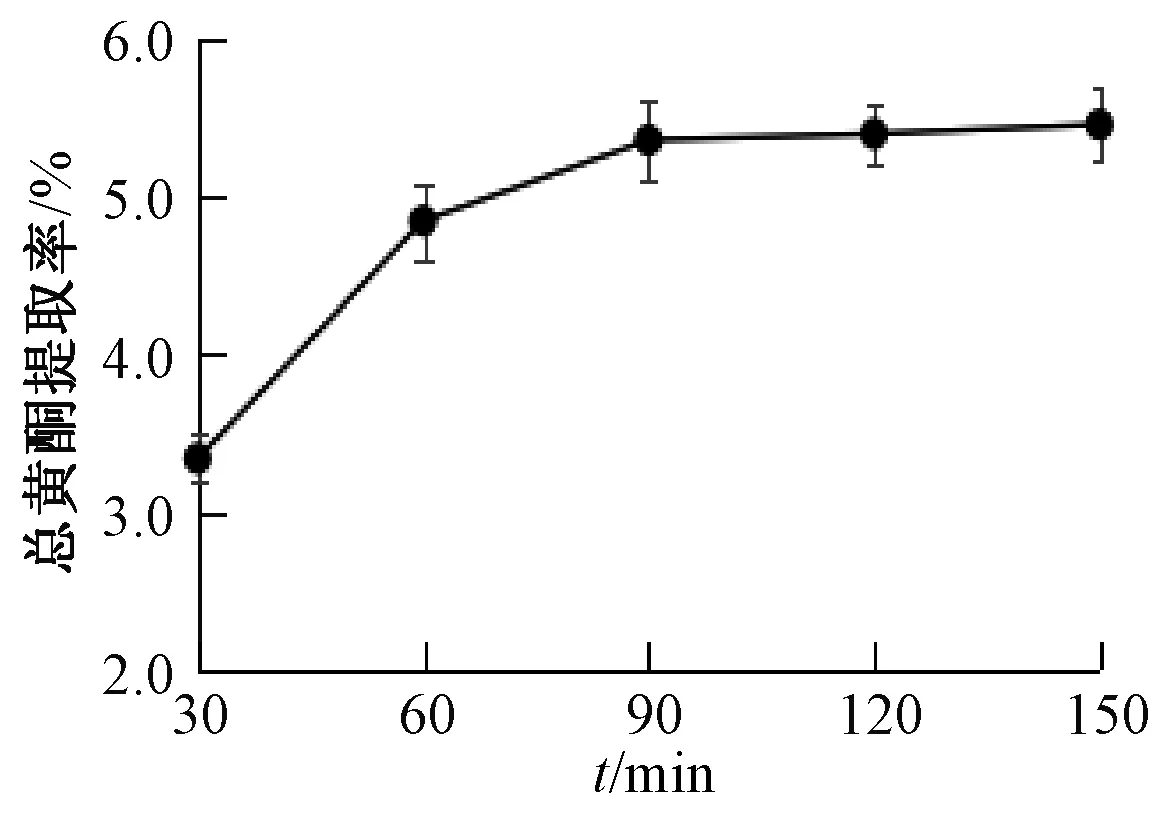
圖4 提取時間對總黃酮提取率的影響
3.2 響應面法 采用Design-Expert 8.0.6軟件安排試驗,結果見表2,方差分析見表3。
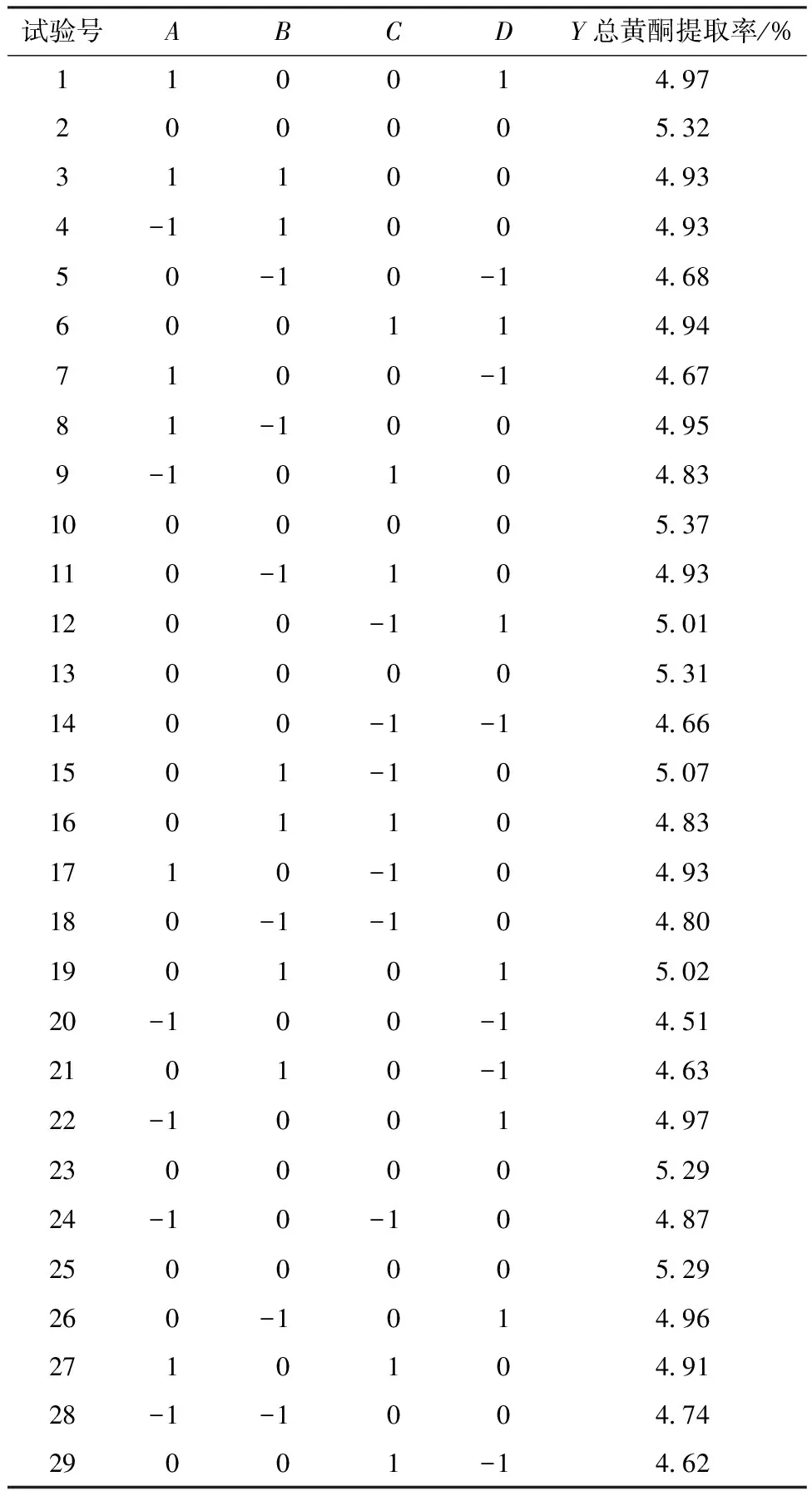
表2 試驗設計與結果

表3 方差分析


注:左圖為三維曲面圖,右圖為等高線圖。
通過Design-Expert 8.0.6軟件,得到最優工藝為乙醇體積分數 60.53%,液料比15.53∶1,提取溫度69.15 ℃,提取時間98.77 min,總黃酮提取率為5.35%,而在實際操作中將其調整為乙醇體積分數61%,液料比16∶1,提取溫度69 ℃,提取時間99 min。再進行3批驗證試驗,測得總黃酮平均提取率為5.28%,與預測值5.35%接近(相對誤差為1.3%),表明該工藝穩定可靠。
3.3 體外生物活性
3.3.1 抗XOD活性 如圖6所示,總黃酮和別嘌醇對XOD均具有抑制作用,并呈顯著的劑量依賴關系,IC50為29.22 μg/mL,約為別嘌呤醇的1/22(1.369 μg/mL)。

圖6 總黃酮對XOD的抑制率
3.3.2 抗氧化活性
3.3.2.1 清除DPPH自由基 如圖7所示,總黃酮具有良好的DPPH自由基清除活性,并呈顯著的劑量依賴關系,IC50為0.127 7 mg/mL,接近維生素C(0.101 6 mg/mL)。
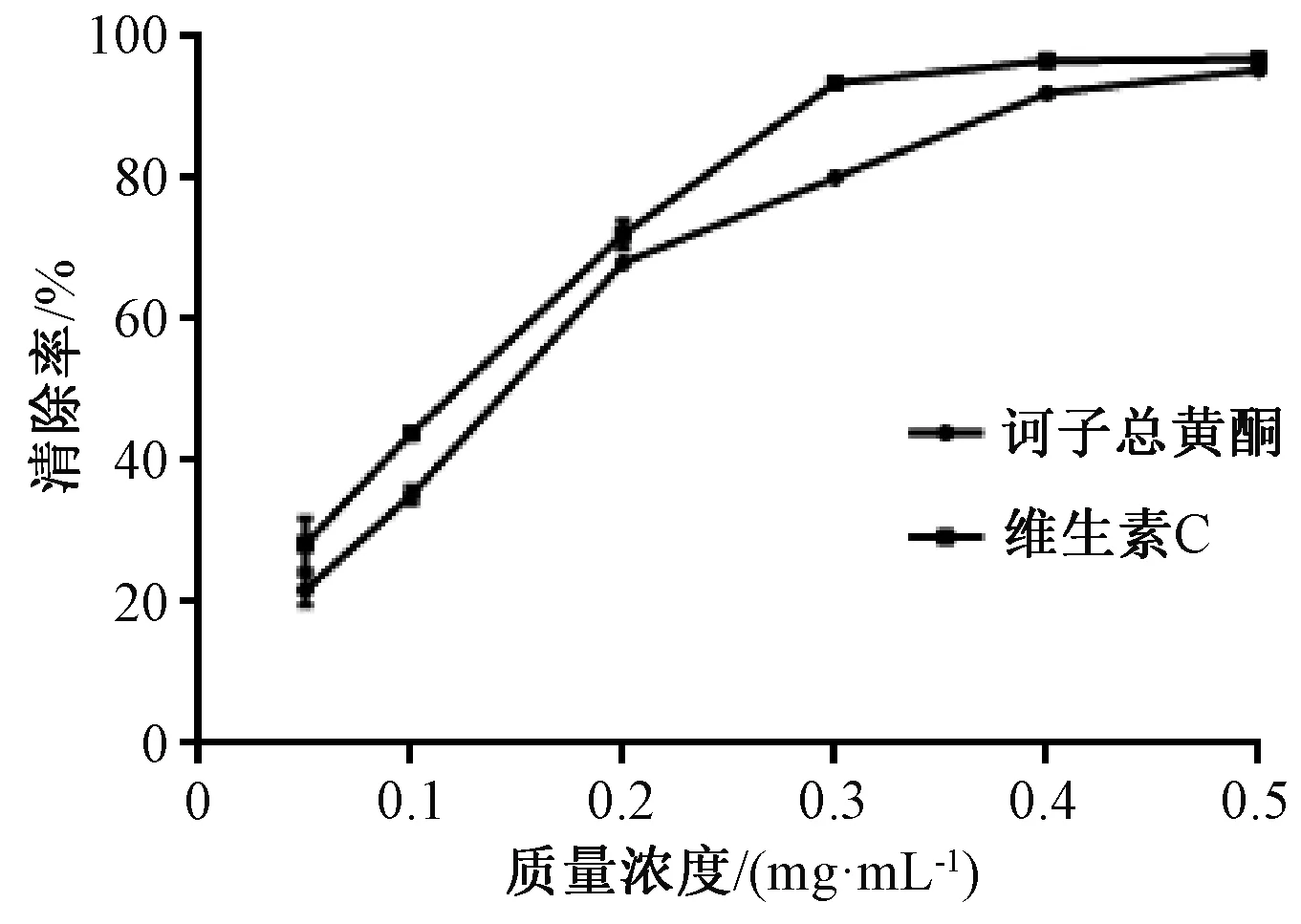
圖7 總黃酮對DPPH自由基的清除率
3.3.2.2 清除羥自由基 如圖8所示,總黃酮具有良好的羥自由基清除活性,并呈顯著的劑量依賴關系,IC50為0.518 8 mg/mL,接近維生素C(0.272 8 mg/mL)。

圖8 總黃酮對羥自由基的清除率
3.3.2.3 清除亞硝酸根離子 如圖9所示,隨著總黃酮質量濃度增加,它對亞硝酸根離子的清除率明顯升高,在200 μg/mL時最高,并在一定范圍內呈量效關系,IC50為42.78 μg/mL,略低于維生素C(11.12 μg/mL)。
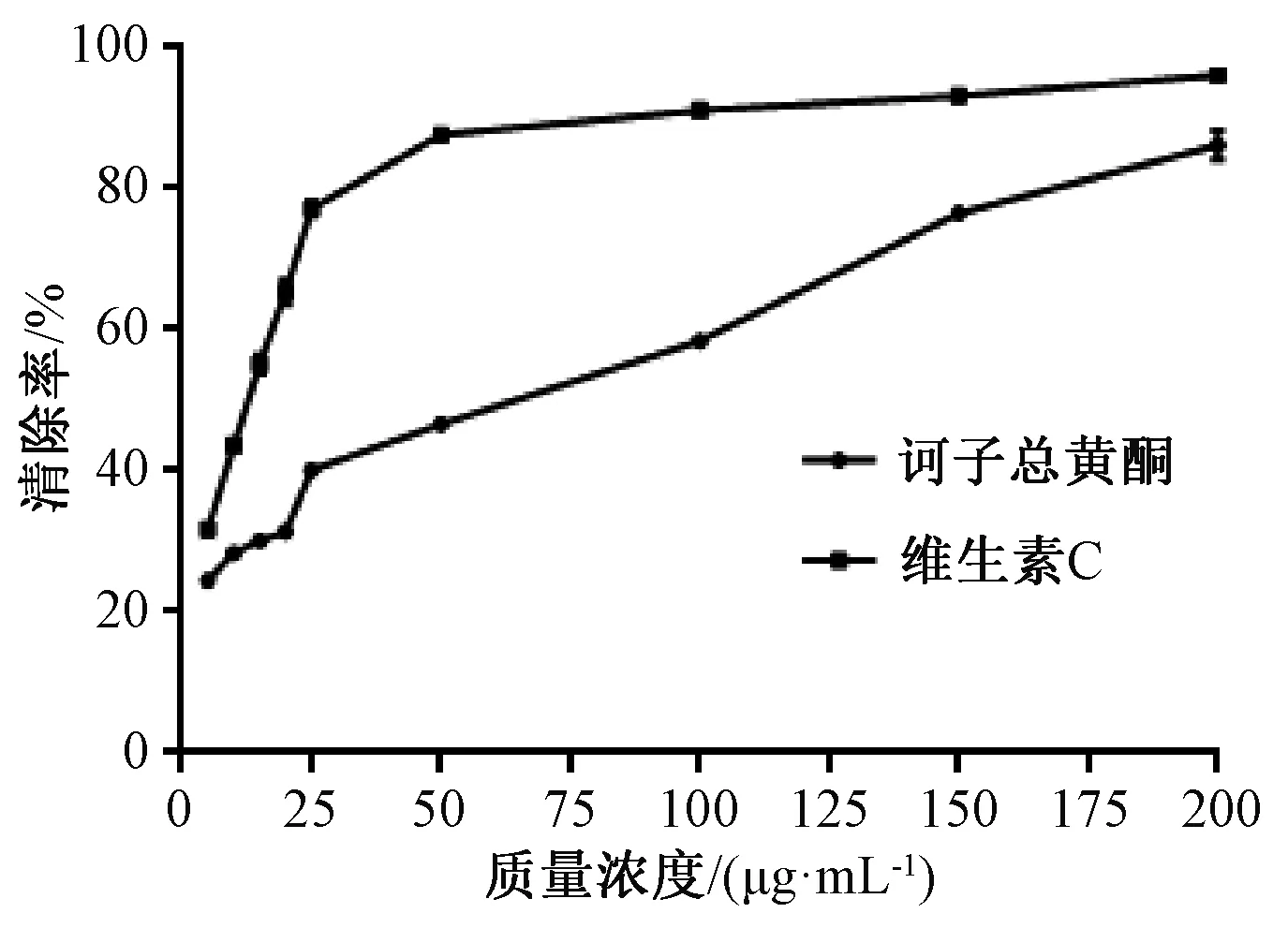
圖9 總黃酮對亞硝酸根離子的清除率
4 討論
于姝燕等[20]采用基于正交設計的微波輔助提取技術來優化訶子總黃酮的提取工藝,但由于微波萃取設備存在微波泄露的風險,導致應用受限,而且未對所提取總黃酮的活性進行評價。因此,本實驗基于回流提取方式,選擇響應面分析法用于優化訶子總黃酮提取工藝參數,該方法在中藥有效成分的提取分離領域中已有廣泛應用[21-22],相比于正交試驗或均勻試驗,它具有試驗次數少、周期短、精度好、預測性強、能研究幾種因素之間的交互作用、準確度高等優點[23]。
體外活性實驗結果表明,在3.75~150 μg/mL質量濃度范圍內,訶子總黃酮對XOD的抑制作用呈劑量依賴關系,IC50為 29.22 μg/mL,表明該成分具有體外抗XOD活性,可為尋找相關天然抑制劑提供依據。此外,訶子總黃酮還顯示出很強的DPPH自由基清除能力(接近于維生素C)及較好的清除羥自由基、亞硝酸根離子能力,其中亞硝酸根(NO2-)是生成N-二甲基亞硝胺(NDMA)的前體,而NDMA具有較強的致突變和致癌性, 對人體健康會產生潛在的危害。因此,后期可將訶子作為潛在的預防高尿酸血癥藥物和抗氧化劑進行深入研究。
猜你喜歡
四川蠶業(2021年2期)2021-03-09 03:15:32
四川蠶業(2021年3期)2021-02-12 02:38:46
中成藥(2018年11期)2018-11-24 02:57:00
中成藥(2017年8期)2017-11-22 03:19:40
中成藥(2017年10期)2017-11-16 00:50:13
中成藥(2017年4期)2017-05-17 06:09:50
哈爾濱醫藥(2016年1期)2017-01-15 13:43:16
天然產物研究與開發(2016年11期)2016-06-15 20:29:17
湖南師范大學自然科學學報(2015年1期)2015-02-27 14:50:04
安徽醫藥(2014年12期)2014-03-20 13:15:15
