基于HPLC指紋圖譜建立醫院紫草藥材驗收標準*
2021-12-01 07:16:52華國棟
中國藥業 2021年22期
關鍵詞:中藥
于 典,黃 磊△,華國棟,何 婷,鞏 穎,楊 維
(1.北京中醫藥大學東方醫院藥學部,北京 100078; 2.北京中醫藥大學東直門醫院藥學部,北京 100700)
對中藥功效的深入研究發現,單一的活性成分或指標性成分均難以反映中藥的真偽和質量[1]。通用現代的分析技術分析中藥和中藥復方,能為鑒別中藥材的真偽和質量提供有效參考[2],其中建立指紋圖譜是評價藥品質量的關鍵環節和基礎[3]。指紋圖譜的參照物質多選擇藥材中易獲取的主要活性成分或指標性成分,以考察藥材圖譜穩定性及輔助辨別圖譜重復性。若臨床藥效未能取得確切關聯,則指紋圖譜的參照物將起到辨別和評價藥材圖譜特征的引導作用[4]。萘醌類、苯醌類、黃酮類、生物堿類及多糖類物質是藥用紫草的主要活性成分,其中萘醌類化合物的抗癌作用和較強的脂溶性特征是當前紫草研究的熱點,左旋紫草素、乙酰紫草素、去氧紫草素、β-β′二甲基丙烯酰紫草素、β-羥基異戊酰紫草素、去氫阿卡寧和β-羥基異戊酰阿卡寧等物質也均是紫草的主要物質成分[5]。本研究中選取左旋紫草素、乙酰紫草素、去氧紫草素、β-β′-二甲基丙烯酰紫草素為參照物,建立了紫草藥材的高效液相色譜(HPLC)指紋圖譜。現報道如下。
1 儀器與試藥
1.1 儀器
Waters e2695型高效液相色譜儀(美國Waters公司);BSA124S型分析天平(北京賽多利斯科學儀器有限公司,精度為0.0001 g)。
1.2 試藥
左旋紫草素對照品(中國食品藥品檢定研究院,批號為110769-200506,含量≥95%);乙酰紫草素對照品(上海士鋒生物技術有限公司,批號為15062715,含量≥98%);去氧紫草素對照品(北京邁瑞達科技有限公司,批號為19072541,含量≥98%);β-β′-二甲基丙烯酰紫草素對照品(上海源葉生物科技有限公司,批號為Y02A9H67135,含量≥98%);紫草(批號分別為ZC-2020001和ZC2020002的來自新疆哈密,批號分別為ZC-2020003和ZC-2020004的來自新疆阿勒泰,批號分別為ZC-2020005和ZC-2020006的來自新疆喀什,批號分別為ZC-2020007和ZC-2020008的來自新疆烏魯木齊,批號分別為ZC-2020009和ZC-2020010的來自尼泊爾);甲醇、乙腈均為色譜純,磷酸為分析純,水為純化水。
2 方法與結果
2.1 色譜條件
色譜柱:Shimadzu VP-ODS C18柱(250 mm×4.6 mm,5μm);流動相:乙腈-0.3%磷酸水溶液(梯度洗脫,洗脫程序見表1);流速:1.0 mL/min;檢測波長:274 nm;柱溫:25℃;進樣量:10μL。

表1 梯度洗脫程序Tab.1 Gradient elution program
2.2 溶液制備
稱取左旋紫草素對照品、乙酰紫草素對照品、去氧紫草素對照品、β-β′-二甲基丙烯酰紫草素對照品各10.00 mg,精密稱定,分別置10 mL容量瓶中,分別以流動相制成質量濃度為0.7,1.0,1.5,1.0 mg/mL的對照品溶液;各精密量取1 mL,置同一100 mL容量瓶中,加流動相定容,搖勻,制成混合對照品溶液。取紫草樣品粉末(過60目篩)0.5 g,精密稱定,加入6倍量的乙醇溶液于具塞錐形瓶中,室溫下密封、遮光。冷浸提取后,過濾,并收集濾液,制成濃縮、干燥的浸膏,加10 mL甲醇使溶解,冷卻至室溫,作為供試品溶液。
2.3 方法學考察
系統適用性試驗:精密量取2.2項下混合對照品溶液、供試品溶液各適量,按2.1項下色譜條件進樣測定,記錄色譜圖。結果供試品溶液色譜中,在與混合對照品溶液色譜相應位置有吸收峰,理論板數按左旋紫草素、乙酰紫草素、去氧紫素、β-β′-二甲基丙烯酰紫草素峰計均大于3000,分離度均大于1.5,基線分離良好。詳見圖1。

圖1 高效液相色譜圖1.L-shikonin 2.acetylshikonin 3.deoxyshikonin 4.β-β′-dimethylacryl shikonin A.Mixed reference solution B.Test solutionFig.1 HPLC chromatograms
精密度試驗:取紫草藥材樣品,按2.2項下方法制備供試品溶液,按2.1項下色譜條件進樣測定6次,記錄相對保留時間及色譜峰面積。結果的RSD均小于1.1%(n=6),表明方法精密度良好。
穩定性試驗:2.2項供試品溶液,室溫下放置0,2,4,8,12,18,24,32,40,48 h時按2.1項下色譜條件分別進樣測定,記錄相對保留時間及色譜峰面積。結果的RSD均小于2.1%(n=10),表明供試品溶液室溫下48 h內穩定。
重復性試驗:取同一批次紫草藥材樣品6份,按2.2項下方法制備供試品溶液,按2.1項下色譜條件進樣測定,記錄相對保留時間及色譜峰面積。結果的RSD均小于1.4%(n=6),表明方法重復性良好。
耐用性試驗:使用不同型號的儀器、不同批次、不同柱溫(28,30,32℃)的色譜柱對同一批次紫草藥材樣品進行測定,計算其相似度。結果表明,在一定波動范圍內,各共有峰的分離度無明顯差異,說明指紋圖譜測定方法的耐用性良好。
2.4 指紋圖譜的建立與評價
2.4.1 紫草指紋圖譜的建立
選擇10批次紫草藥材,分別按上述建立的指紋圖譜測定方法進行檢測,采用“中藥色譜指紋圖譜相似度評價系統”(2012.0版)擬合生成對照指紋圖譜,以峰面積>12.5%的10號峰(去氧紫草素)為對照峰,0.5%<峰面積<12.5%的分別為第1~9號和第11~14號峰,這14個峰的面積之和占共有峰總面積的90%,詳見圖2。10批藥材對照指紋圖譜見圖3。

圖2 10批藥材疊加指紋圖譜Fig.2 Superimposed HPLC fingerprints of 10 batches of Arenbiae Radix
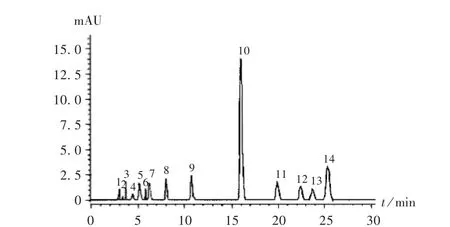
圖3 10批藥材對照指紋圖譜Fig.3 Reference fingerprint of 10 batches of Arenbiae Radix
2.4.2 相似度評價
采用“中藥色譜指紋圖譜相似度評價系統”(2012.0版)建立共有模式,對圖譜數據的相關參數進行自動匹配,生成對照圖譜S,計算10批紫草藥材提取物圖譜與對照圖譜間的相似度。結果多超過0.7,見表2。

表2 10批不同產地紫草的HPLC指紋圖譜相似度Tab.2 Similarity of HPLC fingerprints of 10 batches of Arnebiae Radix from different producing areas
3 討論
當前市場紫草品種良莠不齊,2020年版《中國藥典(一部)》對藥用紫草的描述特征不鮮明,且在紫草指紋圖譜方面尚未建立對應的質量規范標準。紫草的市場來源較復雜,實際流通的并非僅藥典規定的新疆紫草和內蒙古紫草,還有更多不同的紫草品種無法通過《中國藥典》的性狀描述準確鑒別[6]。不同的種植地點、采收期、貯藏條件、運輸條件等均會對藥材產生較大影響,導致現在市場紫草藥材的質量難以控制[7]。
為了規范新疆紫草市場,應建立有效的含量測定方法,保證紫草藥材質量的有效性和安全性[8]。指紋圖譜為全面說明中藥化學成分特點的一種新技術,可表征中藥樣本的特征成分[9]。中藥指紋圖譜為綜合性鑒別方法,其應用范圍是鑒別真偽,評價中藥的質量均勻性和穩定性[10-11]。采集藥材組的指紋圖譜,弄清中藥材化學成分的多樣性和復雜性,是科學合理地控制中藥材質量的有效途徑[12-13]。本研究結果顯示,樣品指紋圖譜和對照藥材指紋圖譜中所含化學成分基本相同,但不同市場圖譜中特征峰數量和各特征峰面積均存在較大差異,表明新疆紫草萘醌提取物在不同來源的組成相似,但含量差異大。
本研究中建立的紫草指紋圖譜用于紫草藥材的驗收標準,即已明確該藥材為紫草的前提下,用指紋圖譜控制紫草的入院質量,目的是用于控制入院紫草藥材的質量均一性及保證臨床療效的均一性。且本研究建立的指紋圖譜雖僅選擇了2個大類產區、5個小類產區,指紋圖譜的色譜峰選擇受到一定限制,但更細化的區分則有利于研究的深入開展。
猜你喜歡
中老年保健(2021年5期)2021-12-02 15:48:21
中老年保健(2021年4期)2021-12-01 11:19:40
中老年保健(2021年4期)2021-08-22 07:08:32
中國現代中藥(2020年10期)2020-12-16 08:53:18
金橋(2020年7期)2020-08-13 03:07:00
基層中醫藥(2020年12期)2020-07-22 06:34:38
中國現代中藥(2020年4期)2020-06-10 09:56:34
基層中醫藥(2018年6期)2018-08-29 01:20:20
長春中醫藥大學學報(2017年1期)2017-04-16 05:56:49
肝博士(2015年2期)2015-02-27 10:49:49
