三重real-time PCR檢測副溶血弧菌主要毒力基因
2021-12-03 09:25:44雷舒文上官文丹鐘青萍
食品科學(xué) 2021年22期
陳 松,雷舒文,上官文丹,劉 丹,鐘青萍
(華南農(nóng)業(yè)大學(xué)食品學(xué)院,廣東省食品質(zhì)量與安全重點實驗室,廣東 廣州 510642)
副溶血弧菌(Vibrio parahaemolyticus)是嗜鹽性的革蘭氏陰性菌,廣泛存在于近海岸水域中。海產(chǎn)品中如魚、蝦、貝類等常攜帶副溶血弧菌[1-2]。食用被致病性副溶血弧菌污染的食品會導(dǎo)致急性腸胃炎,出現(xiàn)腹瀉、惡心、嘔吐、發(fā)熱和腹部痙攣等癥狀[3-4]。據(jù)我國食品安全風險評估中心統(tǒng)計,由副溶血弧菌引起的食物中毒人數(shù)每年可達到495萬 人[5]。此外,副溶血弧菌引起的食物中毒事件在美國、日本、韓國、西班牙等地也常有報道,是一種全世界范圍內(nèi)的主要食源性致病菌[6-8]。
盡管副溶血弧菌對食品安全造成了巨大威脅,但并不是所有的菌株都有致病性,僅有部分副溶血弧菌會引起食源性疾病的爆發(fā)[9]。因此,建立一種快速、靈敏、準確,并能區(qū)分致病性及非致病性副溶血弧菌的檢測方法具有重要意義。研究發(fā)現(xiàn),致病性及非致病性副溶血弧菌都攜帶有編碼不耐熱溶血毒素(thermolabile hemolysin,TLH)的基因(tlh),只有攜帶編碼耐熱直接溶血毒素(thermostable direct hemolysin,TDH)的基因(tdh)和/或編碼耐熱相關(guān)溶血素(thermostable related hemolysin,TRH)的基因(trh)的菌株具有強致病力,而trh的2 個亞型(trh1和trh2)在序列上存在一定的差異[10]。位于trh上游編碼尿素酶的ureR基因高度保守,且與trh具有連鎖關(guān)系,因此以ureR代替trh作為檢測靶標可以有效規(guī)避序列差異的問題[11]。
目前,傳統(tǒng)培養(yǎng)方法仍是檢測副溶血弧菌的公認且可靠的方法,其優(yōu)點是結(jié)果較為準確,但該方法操作比較繁瑣,耗時較長,整個檢測流程一般需要4~7 d,不能滿足快速檢測的要求[12]。聚合酶鏈式反應(yīng)(polymerase chain reaction,PCR)由于其反應(yīng)時間短、結(jié)果準確、靈敏度高等優(yōu)點已廣泛應(yīng)用于食源性致病微生物的檢測中[13]。常規(guī)PCR每次反應(yīng)只能針對一個靶標,且不能進行定量分析,而多重實時PCR(real-time PCR)不僅能在一次反應(yīng)中檢測多個靶標,且彌補了傳統(tǒng)PCR不能定量的缺點[14-15]。本研究針對副溶血弧菌的特異性基因tlh,毒力基因tdh和ureR設(shè)計引物及探針,建立一種快速定量檢測副溶血弧菌毒力因子的三重real-time PCR方法,該方法能準確區(qū)分致病性及非致病性的副溶血弧菌,為副溶血弧菌風險監(jiān)測及食品安全評估提供了參考依據(jù)。
1 材料與方法
1.1 材料與試劑
副溶血弧菌ATCC 17802購于廣東省微生物研究所微生物菌種保藏中心;副溶血弧菌標準菌株J5421由中國軍事醫(yī)學(xué)科學(xué)院微生物流行病研究所贈送,其余8 株副溶血弧菌分離菌株和22 株非副溶血弧菌(表1),由華南農(nóng)業(yè)大學(xué)微生物檢測實驗室保存,所有菌株均于-80 ℃保存。

表1 實驗菌株Table 1 Bacterial strains used in this study
硫代硫酸鹽檸檬酸鹽膽鹽蔗糖(thiosulfate citrate bile salts sucrose,TCBS)瓊脂培養(yǎng)基、胰蛋白胨大豆肉湯(trypticase soy broth,TSB) 廣東環(huán)凱微生物科技有限公司;細菌基因組DNA提取試劑盒 北京索萊寶科技有限公司;牛血清蛋白(bovine serum albumin,BSA)、TaqDNA聚合酶、Mg2+、dNTP 生工生物工程(上海)股份有限公司。
1.2 儀器與設(shè)備
SW-CJ-1B超凈工作臺 蘇州凈化設(shè)備有限公司;Forma Class II A2生物安全柜 美國Thermo公司;X3FR高速冷凍離心機、NanoDrop 2000c超微量分光光度計賽默飛世爾科技(中國)有限公司;CFX96 TouchTM熒光定量PCR儀 伯樂生命醫(yī)學(xué)產(chǎn)品(上海)有限公司。
1.3 方法
1.3.1 菌株的培養(yǎng)
副溶血弧菌在無菌條件劃線于TCBS瓊脂培養(yǎng)基上,37 ℃培養(yǎng)12 h,挑取藍綠色單菌落于含3% NaCl的TSB液體培養(yǎng)基中37 ℃、150 r/min培養(yǎng)16 h;非副溶血弧菌劃線于LB瓊脂培養(yǎng)基中,37 ℃培養(yǎng)12 h,形成清晰單菌落后挑取單菌落于LB液體培養(yǎng)基中37 ℃、150 r/min培養(yǎng)16 h。
1.3.2 模板DNA的制備
利用細菌基因組DNA提取試劑盒制備模板DNA,具體步驟按照試劑盒說明書進行。提取后的DNA以超微量分光光度計測定其濃度,并保存于-20 ℃。
1.3.3 引物及探針的設(shè)計與合成
以副溶血弧菌tlh、tdh、ureR基因為靶基因,利用Primer Premier 5.0軟件進行引物探針設(shè)計并由生工生物工程(上海)股份有限公司合成。引物和探針序列及擴增產(chǎn)物見表2。
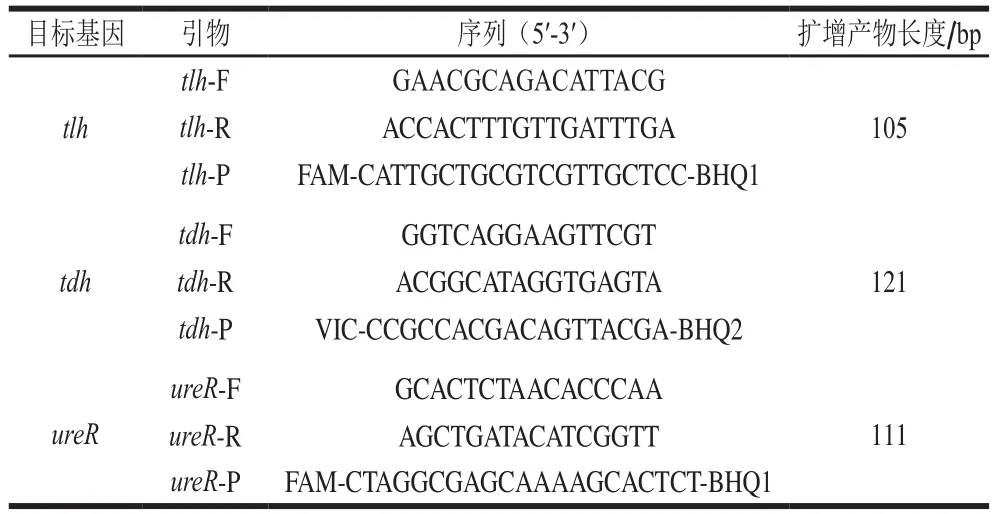
表2 引物和探針序列Table 2 Sequences of the primers and probes used in this study
1.3.4 引物及探針的特異性驗證
以10 株副溶血弧菌菌株和22 株非副溶血弧菌的DNA為模板進行real-time PCR。反應(yīng)體系(20 μL)為:Mix1(K+20 mmol/L,NH4+10 mmol/L,dNTP 120 μmol/L,Mg2+2.5 mmol/L,BSA 0.04 mg/mL,Tris-HCl 50 mmol/L,TaqDNA聚合酶0.1 U/mL,甘油2.50%)10 μL,引物0.80 μmol/L,探針0.50 μmol/L,模板2 μL,補充無菌蒸餾水至20 μL。反應(yīng)條件:95 ℃預(yù)變性10 min;95 ℃變性15 s;53.9 ℃退火15 s;62.4 ℃延伸45 s;每個循環(huán)結(jié)束后采集熒光信號,共45 個循環(huán)。
1.3.5 三重real-time PCR方法的建立及條件優(yōu)化
優(yōu)化前三重real-time PCR體系(20 μL)為:Mix2(K+10 mmol/L,NH4+20 mmol/L,dNTP 180 μmol/L,Mg2+3 mmol/L,BSA 0.04 mg/mL,Tris-HCl 50 mmol/L,TaqDNA聚合酶0.1 U/mL,甘油2.50%)10 μL,tlh、tdh和ureR引物各0.50 μmol/L,tlh、tdh和ureR探針各0.50 μmol/L,模板2 μL,補無菌蒸餾水至20 μL。反應(yīng)條件同1.3.4節(jié)。
1.3.5.1 引物濃度的優(yōu)化
通常,多重PCR引物分配不當會導(dǎo)致目標DNA擴增不均勻,所以在反應(yīng)中必需改變不同引物的比例,目的是增強“弱”位點的擴增和減弱“強”位點的擴增,使得各個目的基因擴增均勻。引物濃度的優(yōu)化分為2 組,第1組為tdh0.15 μmol/L、tlh0.15 μmol/L、ureR分別為0.20、0.40、0.60、0.80、1.00 μmol/L;第2組為tdh0.25 μmol/L、tlh0.25 μmol/L、ureR分別為0.20、0.40、0.60、0.80、1.00 μmol/L。除引物濃度外,其他條件保持不變,根據(jù)優(yōu)化前的反應(yīng)體系及反應(yīng)條件進行real-time PCR優(yōu)化實驗。
1.3.5.2 探針濃度的優(yōu)化
探針的濃度分別設(shè)置為0.50、0.75、1.00、1.25 μmol/L,除探針濃度外,其他條件不變,根據(jù)優(yōu)化前的反應(yīng)體系及反應(yīng)條件進行real-time PCR優(yōu)化實驗,探究探針濃度的影響,選擇合適的探針濃度。
1.3.6 三重real-time PCR靈敏度的評價
將已知濃度的10 倍梯度稀釋的副溶血弧菌及大腸桿菌菌懸液按照試劑盒方法分別提取DNA,并且分為2 組進行實驗,對照組模板為副溶血弧菌(1.8×102~1.8×107拷貝/mL),實驗組模板為副溶血弧菌(1.8×102~1.8×107拷貝/mL)和背景細菌(濃度為106拷貝/mL的大腸桿菌)的混合物,分別進行real-time PCR。根據(jù)優(yōu)化后的體系進行real-time PCR,反應(yīng)程序同1.3.4節(jié)。以real-time PCR結(jié)果為縱坐標,平板計數(shù)的結(jié)果為橫坐標,繪制標準曲線。
1.3.7 以完整菌細胞的懸液和gDNA為模板的檢測
為了確定以完整菌細胞的懸液作為模板對real-time PCR擴增產(chǎn)生的影響,分別將完整菌細胞的懸液和gDNA進行real-time PCR擴增。以gDNA為模板時反應(yīng)程序按照1.3.4節(jié)進行。以完整菌細胞的懸液為模板時,將1 μL菌細胞懸液代替gDNA進行反應(yīng),預(yù)變性時間分別設(shè)置為10、20、30 min,進入循環(huán)95 ℃變性15 s;53.9 ℃退火15 s;62.4 ℃延伸45 s;每個循環(huán)結(jié)束后采集熒光信號,共45 個循環(huán)。
1.4 數(shù)據(jù)處理
每次實驗設(shè)置3 個平行復(fù)孔,并進行2 次重復(fù)實驗。實驗數(shù)據(jù)應(yīng)用Graphpad Prism 8軟件進行平均值和標準偏差的計算。
2 結(jié)果與分析
2.1 引物及探針的特異性驗證
以10 株副溶血弧菌和22 株非副溶血弧菌對設(shè)計的3 對引物進行特異性驗證,結(jié)果如表3所示。結(jié)果表明:所有的副溶血弧菌均為tlh陽性,分別具有tdh或ureR基因的菌株僅對tdh或ureR熒光探針顯示陽性信號。所有非目標菌株的結(jié)果均呈陰性,因此該方法具有很高的特異性。

表3 引物及探針特異性結(jié)果Table 3 Specificity of the primers and probes
2.2 三重real-time PCR方法的建立及條件優(yōu)化
2.2.1 引物濃度的優(yōu)化
ureR由HEX標記,tdh和tlh由FAM標記。如圖1a所示,當FAM通道引物濃度不變,HEX通道引物濃度上升時,F(xiàn)AM通道Ct值上升,HEX通道Ct值下降,即FAM通道引物的擴增效率降低,HEX通道引物的擴增效率升高,當ureR濃度為0.80 μmol/L時,此時三者的擴增效率得到平衡。如圖1b所示,隨著HEX通道引物濃度升高,F(xiàn)AM和HEX通道的變化趨勢與圖1a一致,當ureR濃度為0.80 μmol/L時,三者的擴增效率相對平衡。對比圖1a、b可知,當FAM通道引物濃度的升高,HEX通道的引物濃度不變時,F(xiàn)AM通道Ct值降低,HEX通道的Ct值升高,但FAM通道引物濃度的增加,沒有改善擴增效率,表明在三重real-time PCR中,引物間相互影響,引物tdh0.15 μmol/L、tlh0.15 μmol/L、ureR0.8 μmol/L為最優(yōu)組合。
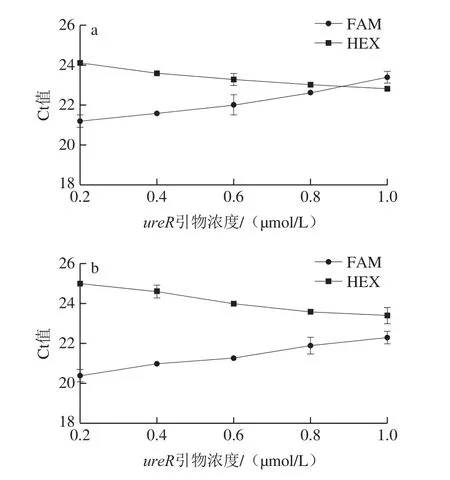
圖1 引物濃度對三重real-time PCR擴增的影響Fig.1 Effects of primer concentration on Ct value of triplex real-time PCR
2.2.2 探針濃度的優(yōu)化
如圖2可知,在三重real-time PCR中,探針濃度對FAM和HEX通道的擴增影響一致,隨著探針濃度的增加,Ct值均增大,在探針濃度為0.50 μmol/L時最優(yōu)。
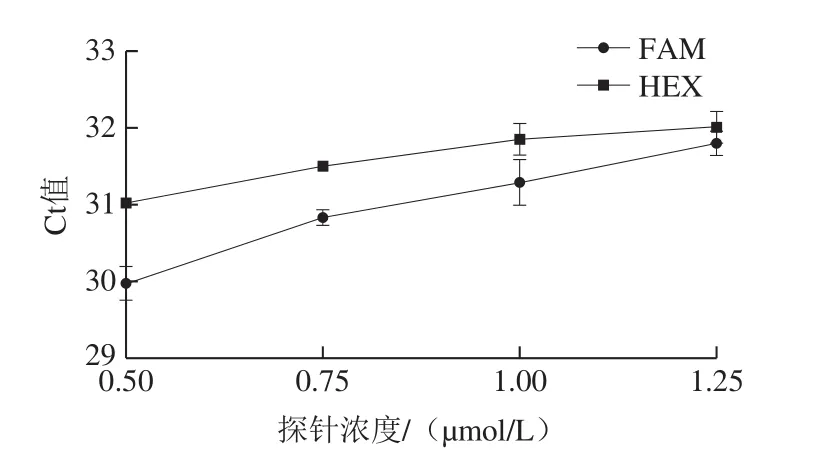
圖2 探針濃度對三重real-time PCR擴增的影響Fig.2 Effect of probe concentration on Ct value of triplex real-time PCR
2.3 三重real-time PCR靈敏度
不同濃度(1.8×102~1.8×107拷貝/mL)副溶血弧菌模板的三重real-time PCR擴增曲線如圖3所示。以DNA濃度對數(shù)值(x)與Ct值(y)繪制標準曲線,在無背景細菌存在下,F(xiàn)AM和HEX通道的線性方程分別為y=-3.564 3x+45.367和y=-3.456 3x+45.77,R2分別為0.998 5和0.997 5,擴增效率分別為91%和95%;在濃度為106拷貝/mL的大腸桿菌為背景下,F(xiàn)AM和HEX通道的線性方程分別為y=-3.484 2x+45.226和y=-3.317 1x+44.923,R2分別為0.997 4和0.999 5,擴增效率分別為94%和100%。DNA濃度對數(shù)值與Ct值均呈良好的線性關(guān)系,與背景細菌的存在無關(guān),檢出限為1.8×102拷貝/mL。
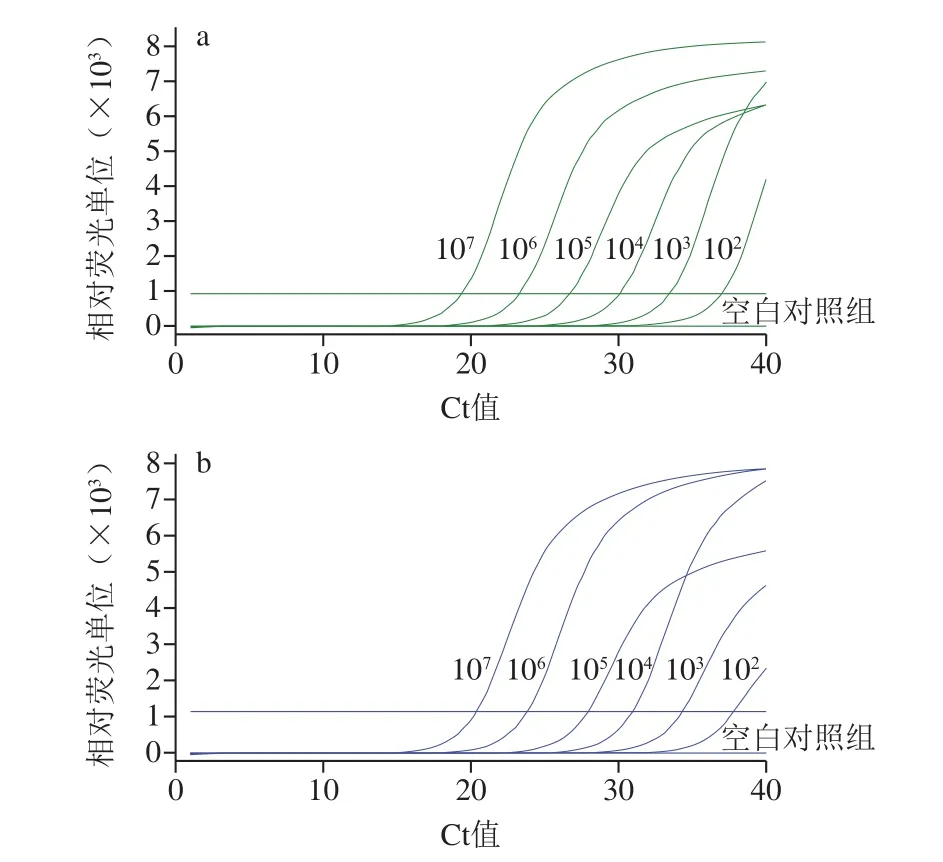
圖3 real-time PCR的FAM通道(a)、HEX通道(b)靈敏度Fig.3 FAM channel (a) and HEX channel (b) sensitivity of real-time PCR
2.4 以完整菌細胞的懸液和gDNA為模板的檢測
如圖4a所示,以完整菌細胞的懸液為模板,當樣品的預(yù)處理時間延長時,Ct值降低,增加到30 min時,擴增檢測效果與以gDNA為模板相比(圖4b),Ct值相差較小(ΔCt<1)。這表明使用完整菌細胞的懸液作為模板與使用gDNA作為模板檢測效果相當,具有準確性和可行性,而且節(jié)省了提取制備模板的時間。
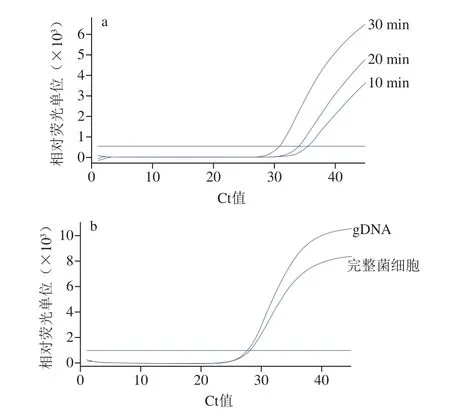
圖4 以完整菌細胞的懸液和gDNA為模板對real-time PCR的影響Fig.4 Influence of intact bacterial cell suspension versus gDNA as template on real-time PCR performance
3 討 論
副溶血弧菌作為一種世界范圍內(nèi)重要的食源性致病菌,具有高度的遺傳多樣性[16]。據(jù)統(tǒng)計,近年我國由副溶血弧菌引起的食物中毒人數(shù)已超過沙門氏菌,躍居首位[17]。因此,建立一種針對副溶血弧菌的準確、快速、靈敏的檢測方法非常必要。本研究中的靶基因tlh具有種的特異性,常被用作副溶血弧菌快速檢測的靶標[18-19];tdh雖然具有5 個不同的亞型(tdh1、tdh2、tdh3、tdh4、tdh5),但其相似度高達97%,具有很高的同源性;而trh1和trh22 個亞型在基因序列上具有一定的差異,同源性僅為84%,使用trh作為檢測靶標可能會在一定程度上導(dǎo)致分類錯誤[20-21]。Okuda等[22]將副溶血弧菌尿素酶陽性(Ure+)分離菌株和陰性(Ure-)分離菌株作為樣本,檢測其攜帶trh基因的概率,結(jié)果發(fā)現(xiàn):在60 株Ure+分離株中,高達98.3%(59/60)菌株攜帶trh基因;而在25 株Ure-分離株中,無trh陽性株,表明trh基因與ure基因呈正相關(guān)。Iida等[23]利用長而準確的PCR檢測發(fā)現(xiàn),trh與ure的間距不超過8.5 kb,表明其在副溶血弧菌上有連鎖關(guān)系,進一步證實了兩者的相關(guān)性。使用高度保守的ureR基因代替trh基因為檢測靶標,可有效規(guī)避因trh基因缺少序列同一性導(dǎo)致分類錯誤的問題。本研究建立的三重realtime PCR方法根據(jù)tlh可區(qū)分副溶血弧菌菌株及非副溶血弧菌菌株,以tdh和ureR判斷副溶血弧菌對強致病性菌株的從屬性,結(jié)果表明建立的方法具有良好的特異性。
隨著副溶血弧菌大流行菌株的出現(xiàn),對副溶血弧菌的關(guān)注度越來越高,同時對副溶血弧菌的檢測方法進行了大量的研究。冼鈺茵等[24]針對副溶血弧菌常見的11 種毒力基因(toxR、Collagenase、toxS、trh、tdh、tlh、ureR、flaA、ompW、aspA、fur)建立了2 套六重PCR檢測體系,均具有良好的特異性,但其不能進行定量分析,且檢出限較高(分別為103、104CFU/mL),而本研究建立的方法能進行準確定量,檢出限低至1.8×102拷貝/mL。Zhong Qingping等[25]建立了疊氮溴化丙錠結(jié)合實時熒光環(huán)介導(dǎo)等溫擴增技術(shù)(loop-mediated isothermal amplification,LAMP)對副溶血弧菌進行檢測,該方法能有效檢測處于活的不可培養(yǎng)態(tài)的副溶血弧菌,在純培養(yǎng)液中檢出限低至6.8 拷貝/mL,在海產(chǎn)品中檢出限為14 拷貝/g,其優(yōu)點為有效區(qū)分死活細胞,檢測時間短,但LAMP對引物要求較高,設(shè)計較為復(fù)雜。高世光等[26]建立了基于Taqman探針檢測副溶血弧菌tlh、trh毒力基因的雙重real-time PCR方法,該方法特異性好,檢出限低至20 CFU/mL,而本研究建立的方法能在一次反應(yīng)中檢測出tlh、tdh、ureR3 個毒力基因,與該方法相比,本研究建立的方法可對副溶血弧菌菌株進行更精準的分類,進一步評估菌株的致病力。萬瑩等[27]針對副溶血弧菌的toxR、trh、tdh建立了三重real-time PCR的檢測方法,toxR作為副溶血弧菌的檢測靶標,與tlh具有相當?shù)撵`敏度及特異性[28-29],而本研究使用ureR而非trh更能避免分類錯誤的問題。此外,本研究直接以完整菌細胞的懸液作為模板可縮短檢測時間而不影響Ct值,方便快捷。隨著微滴數(shù)字PCR(droplet digital PCR,ddPCR)的出現(xiàn),對副溶血弧菌的檢測方法的研究也得到了巨大突破。Lei Shuwen等[30]建立了ddPCR檢測副溶血弧菌tlh、trh、ureR及orf8的四重檢測方法,該方法能夠?qū)Ω比苎【鷿舛确秶?.9×101~3.9×107CFU/mL的實際食品樣品進行準確測定,并能準確鑒定攜帶不同毒力基因的副溶血弧菌,但高價儀器一定程度上限制了該方法的廣泛應(yīng)用。
4 結(jié) 論
本研究建立的三重real-time PCR方法能在一次反應(yīng)中準確、快速、靈敏地定量檢測副溶血弧菌,并能區(qū)分致病性和非致病性副溶血弧菌;在高濃度的背景細菌存在下,該方法仍具有良好的特異性,其檢測的線性范圍為1.8×102~1.8×107拷貝/mL,檢出限為1.8×102拷貝/mL;此外,該方法可以直接將菌液作為模板進行擴增,樣品預(yù)變性時間為30 min時,其Ct值與使用DNA為模板的Ct值相近。本研究建立的方法為食源性致病菌檢測提供了參考依據(jù),具有較好的推廣應(yīng)用價值。
猜你喜歡
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
海峽科技與產(chǎn)業(yè)(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
