基于GC-MS/MS技術測定茶葉中18種多氯聯(lián)苯*
2021-12-16 01:36:08蘇曉明
福建輕紡 2021年12期
蘇曉明
(福建省產(chǎn)品質量檢驗研究院,福建 福州 350002)
茶在我國源遠流長,被譽為世界三大重要加工飲品之一,深受國內(nèi)外消費群體的青睞。隨著茶產(chǎn)品的普及以及產(chǎn)量的日益增加,茶葉中的多氯聯(lián)苯(Polychlorinated biphenyls,PCBs)等持久性有機污染物已成為當下不可忽視且亟需解決的重要問題[1,2]。
多氯聯(lián)苯又稱氯化聯(lián)苯,在化學組成上聯(lián)苯分子上的氫原子被氯原子取代而形成的氯代芳烴類混合[2]。2001年瑞典斯德哥爾摩通過了《關于持久性有機污染物的斯德哥爾摩公約》,PCBs成為首批被列入該公約的受控物質之一[3]。PCBs難溶于水、易溶于有機溶劑,具有高毒性、積聚性、遷移性等特性,易在生物體內(nèi)富集而產(chǎn)生生物放大效應,進入人體后會影響人體的發(fā)育、神經(jīng)系統(tǒng)和免疫系統(tǒng),對人體健康和生態(tài)系統(tǒng)具有嚴重的威脅[4]。目前,對于多氯聯(lián)苯的研究主要針對水體、工業(yè)產(chǎn)品、土壤、沉積物等,而茶葉中PCBs殘留量的分析卻鮮有報道,因此開發(fā)高效、快速、準確的檢測方法成為眾多學者關注的重點。
目前PCBs的檢測方法主要有氣相色譜-電子捕獲檢測器法(GC-ECD)和氣相色譜-質譜法(GCMS)[1,5],前處理技術通常采用加速溶劑萃取、索氏提取等凈化方法,由于PCBs化合物種類繁多、部分樣品基質復雜,操作繁瑣,易造成多氯聯(lián)苯定性定量不準確。氣相色譜-三重四級桿串聯(lián)質譜法(gas chromatography-tandem mass spectrometry,GCMS/MS)[6]是近年來迅速發(fā)展的檢測技術,它采用定時SRM數(shù)據(jù)采集功能,能同時完成組分分離和結構分析,具有準確可靠、分離效率高、分析速度快且能縮短檢測周期、降低成本等優(yōu)點。本研究以鐵觀音茶葉樣品作為研究對象,首次將氣相色譜-三重四級桿串聯(lián)質譜技術應用于茶葉中18種PCBs的分析檢測,為茶葉中PCBs的定量分析和快速監(jiān)測、以及標準方法的制定提供技術支持。
1 材料與方法
1.1 儀器、試劑與材料
試驗所用主要儀器設備詳見表1。

表1 實驗儀器設備一覽表
實驗所用試劑與材料為:無水硫酸鎂(分析純,太倉滬試試劑有限公司);正己烷(分析純,經(jīng)重蒸制得);二氯甲烷(分析純,經(jīng)重蒸制得);N-丙基乙二胺(PSA,粒度4μm,GL Sciences InC);MIPs/SPE(CNWBOND ,1 g/10 mL,德國CNW公司);多壁碳納米管(MWCNTS,粒度5 nm,Agela Technologies)。
實驗用水均為Milli-Q超純水器純化水。18種PCBs標準溶液質量濃度均為100μg/mL(購于美國Accustandard公司),基本信息詳見表2。
1.2 實驗條件
標準溶液及標準曲線配制:
標準中間液:取0.20 mL多氯聯(lián)苯標準品混合物于10 mL容量瓶中,用正己烷定容至10 mL,充分搖勻,配制成18種PCBs標準中間液,-20 ℃冰箱中保存。
標準工作液:取2.50 mL標準中間液于10 mL容量瓶中,用正己烷定容至10 mL,充分搖勻,配制成0.50μg/mL的PCBs標準工作液;使用逐級稀釋的方法,配制成濃度為0.001、0.002、0.01、0.05、0.5μg/mL的標準使用液,于4~8 ℃冰箱中保存待用。
1.3 樣品前處理
1.3.1 樣品提取
取2.00 g粉碎均勻的茶葉粉末,加入1.0 g無水Na2SO4,使用20 mL正己烷-丙酮(9:1,v/v)進行超聲提取20 min,渦旋混勻,而后離心5 min,轉速為10000 r/min,取上清液于試管中,氮吹濃縮近干,正己烷定容至1 mL,待凈化。
1.3.2 提取液凈化
提取液的凈化采用CNWBOND分子印跡柱,依次用5 mL的二氯甲烷和正己烷對柱子進行活化,而后將待凈化液轉移至活化后的分子印跡柱中,待液面降至柱床時,用6 mL正己烷淋洗柱子,流出液用氮吹濃縮至約2 mL后加入QuEchERS凈化包(0.15 g MWCNTS、0.15 g PSA、0.15 g無水硫酸鎂),攪拌均勻,而后離心5 min,轉速為10000 r/min。再用6 mL二氯甲烷洗脫上述分子印跡柱,收集洗脫液與QuEchERS凈化后溶液合并混勻,氮吹濃縮近干,最后用正己烷定容至2.0 mL,過0.22 μm濾膜上機測試。
1.4 氣相色譜-三重四級桿串聯(lián)質譜條件
1.4.1 色譜條件
色譜柱:DB-5ms(30 m×250 μm×0.25 μm);進樣方式:不分流進樣;色譜柱流量:1.0 mL/min;壓力(恒壓):8.0013 psi;進樣口溫度:250 ℃;載氣:氦氣(純度>99.999%);進樣量:1.0 μL。
1.4.2 質譜條件
離子源:電子轟擊離子源(EI);碰撞氣:氮氣(純度>99.999%);離子源溫度:280 ℃;數(shù)據(jù)采集模式:MRM;傳輸線溫度:290 ℃;電離能量:70 eV ;溶劑延遲時間:4 min;18種PCBs的MS/MS分析參數(shù)見表3。
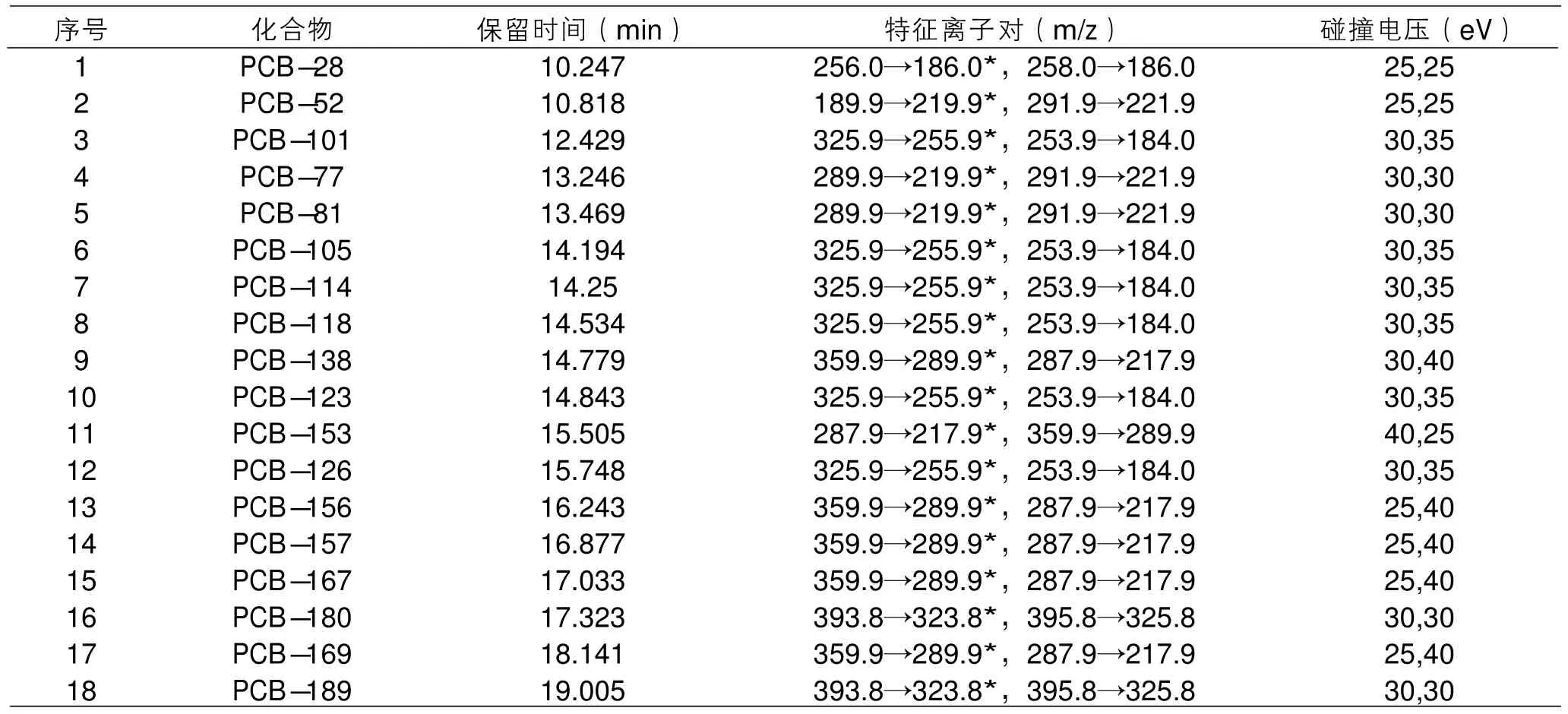
表3 18種PCBs的保留時間、離子對信息和碰撞電壓
2 結果與討論
2.1 前處理條件的優(yōu)化
2.1.1 提取溶劑
為確定最佳的提取溶劑,以鐵觀音茶葉為基質,在0.05 mg/kg的加標水平下系統(tǒng)考查正己烷、甲苯、二氯甲烷、正己烷-丙酮(9∶1,v/v)和正己烷-丙酮(1∶1,v/v)5種提取劑對18種PCBs目標物提取效果的影響。
實驗結果(表4)表明,對于單一提取劑,正己烷相對其他兩種提取劑具有更高的提取率;對于混合提取劑,正己烷-丙酮(9∶1,v/v)、正己烷-丙酮(1∶1,v/v)二者的平均回收率相差不大,但均優(yōu)于直接采用正己烷時的提取效果。由于正己烷-丙酮(1∶1,v/v)萃取液中含有大量色素類雜質,因此,選擇正己烷-丙酮(9∶1,v/v)混合液作為本試驗的提取劑。

表4 提取溶劑對18種PCBs 回收率的影響
2.1.2 提取方式
本實驗以鐵觀音樣品為基質,采用超聲提取法進行前處理,考察不同超聲提取時間(5、10、20、30 min)對目標回收率的差異。結果表明,超聲提取時間由5 min增加至10、20、30 min時,18種多氯聯(lián)苯的回收率逐漸增大。超聲時間為10 min時,18種PCBs的回收率介于56%~75%之間;超聲時間為20 min時增加至90%~103%,超聲時間為30 min時,回收率介于88%~106%之間,該回收率與超聲20 min所得結果并無較大差異,故20 min為最優(yōu)超聲提取時間。
此外,在上述實驗的基礎也比較了不同超聲溫度(60、70、80、90、100 ℃)下,鐵觀音樣品的加標回收率。結果如圖1所示,超聲溫度為70 ℃和80 ℃時,部分高沸點PCBs的回收率相對較;超聲溫度為100 ℃和90 ℃時,大部分多氯聯(lián)苯的回收率相對較高,因此本試驗最終選取100 ℃的超聲處理溫度。
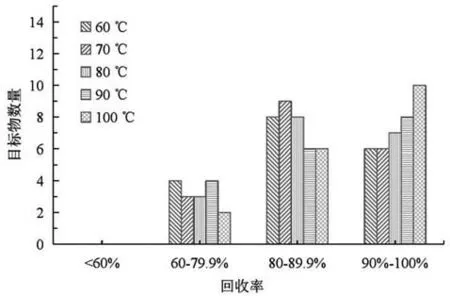
圖1 超聲溫度對18種PCBs 回收率的影響
2.1.3 QuEchERs凈化條件
為保證儀器檢測結果的準確性,需使用凈化劑去除樣品中的色素、兒茶素、茶多酚等干擾雜質。常用的凈化劑有C18、PSA[7]、MWCNTS[8]、Florisil[9]等,其中C18能吸附非極性物質,去除少量色素、甾醇和脂肪等物質;PSA能去除樣品中的有機酸、茶多酚和兒茶素類物質;MWCNTS對色素的吸附效果最佳,而采用其他凈化填料凈化后,溶液均有一定顏色。因此本試驗選擇PSA、C18、MWCNTS為凈化劑,按照1.3節(jié)條件處理鐵觀音樣品,考察了0.20 mg/kg水平加標濃度下的凈化效果,實驗結果發(fā)現(xiàn),單獨采用C18凈化劑時,PCB28、PCB52兩個化合物的吸附比較嚴重,不予采用。
為保證18種多氯聯(lián)苯的回收率以及凈化效果,對吸附劑的用量進行探究。在上述實驗的基礎上,實驗將PSA、MWCNTS混合使用,以加標水平為0.20 mg/kg的鐵觀音為基質,考察吸附劑用量(0.10 g PSA+0.10 g MWCNTS、0.10 g PSA+0.15 g MWCNTS、0.15 g PSA+0.10 g MWCNTS、0.15 g PSA+0.15 g MWCNTS)對18種PCBs回收率的影響。結果如圖2所示,當以0.15 g PSA+0.15 g MWCNTS為凈化劑時,18種多氯聯(lián)苯的回收率在85.4%~105.3%之間,凈化效果最佳。
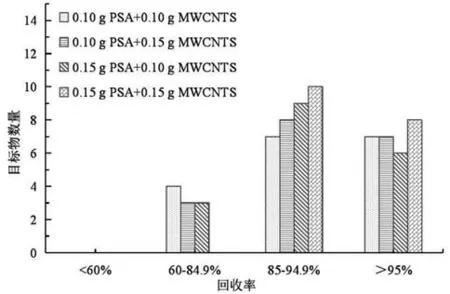
圖2 不同的凈化劑用量對18種PCBs 回收率的影響
2.2 儀器測試參數(shù)
2.2.1 初始柱溫、升溫速率優(yōu)化的選擇
由于多氯聯(lián)苯分子中的氫原子被氯原子取代的方式及數(shù)目不同,理化性質尤其是沸點跨度大,色譜峰形和響應狀況差異明顯。過低的初始柱溫,將使低沸點組分出峰過遲,峰形拖尾,從而影響之后的組分出峰;程序升溫速度太慢,沸點高的組分出峰容易延遲,峰形擴展嚴重。
實驗采用DB-5ms色譜柱,通過下列兩組條件(均為程序升溫),考察升溫程序對樣品色譜分離的影響。條件A:初始溫度60 ℃,保持1 min,而后以40 ℃/min升溫至170 ℃,保持1 min;最后以10 ℃/min升溫到310 ℃,保持5 min。條件B:初始溫度為40 ℃,保持5 min,以40 ℃/min升溫至180 ℃,保持1 min;最后以15 ℃/min升至280 ℃,保持5 min。條件A和條件B兩者均得到良好的分離度,但色譜峰的峰型、峰高響應值存在較明顯的差異,而條件A得到的色譜峰型、峰高響應明顯優(yōu)于條件B,因此本實驗采用條件A作為試驗的色譜條件。
2.2.2 氣相色譜質譜條件的優(yōu)化
為獲得質量數(shù)較大、豐度較高的特征離子來作為目標離子的母離子,需將目標化合物進行全掃描。MRM模式相比于SIM模式具有靈敏度高、專屬性好、抗干擾能力強等特點,是目前最靈敏的MS模式,尤其適用于持久性有機污染物的檢測,且MRM模式能夠有效提高多種多氯聯(lián)苯的分辨率,而SIM模式中存在相同的掃描離子,這對于多種持久性有機污染物的定性/定量檢測帶來困擾,因此本研究采用MRM采集模式。
在2.2.1優(yōu)化的色譜條件下,采用GC-MS/MS全掃描方式,對目標化合物進行分析,18種PCBs均得到了良好的分離,其總離子流色譜圖如圖3所示。
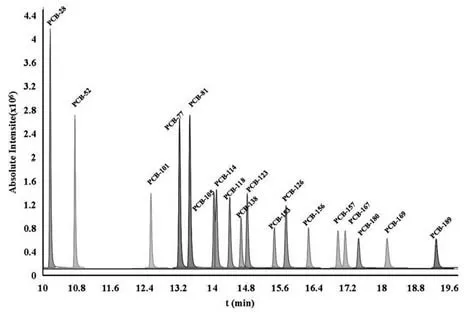
圖3 18種PCBs混合標準溶液的總離子流圖
2.2.3 基質效應
對于痕量檢測來說,基質效應是定量分析中必須考慮的問題。樣品溶液中其他組分改變了目標物的響應值,從而達到基質抑制或增強的效應稱為基質效應[2]。評定基質抑制或增強的主要因素為基質匹配標準曲線斜率和純?nèi)軇藴是€斜率的比值。當比值小于0.9時,為基質抑制效應;當比值介于0.9~1.1之間時,基質效應可以忽略;當比值大于1.1時,為基質增強效應[10]。以空白鐵觀音樣品來探究茶葉中PCBs的基質效應(見表5),其中化合物PCB-189表現(xiàn)出一定程度的基質增強效應,其余組分的斜率比在0.93~1.09之間,基質效應可忽略。因此試驗采用基質匹配標準曲線進行定量,可補償基質效應對試驗結果的影響。
2.3 方法評價
2.3.1 線性范圍、檢出限和定量限
選取已知空白鐵觀音樣品采用1.3節(jié)的方法進行處理,制得基質提取液后,用逐級稀釋的方法制備與1.2節(jié)濃度一致的基質標準系列溶液,分別測定。以每種PCBs的定量離子對的峰面積(y)為縱坐標,質量濃度(x,μg/L)為橫坐標繪制標準曲線,結果見表5。檢測結果顯示樣品中的PCBs在各自線性范圍內(nèi)均呈良好的線性關系,相關系數(shù)R2≥0.999,檢出限為0.1~0.7μg/kg,定量限為0.4~2.3μg/kg,滿足分析需求。

表5 18種多氯聯(lián)苯的線性關系、檢出限、定量限和基質效應
2.3.2 回收率及精密度
以鐵觀音茶葉為基質,在探索最佳的試驗條件下進行加標回收試驗,添加水平為低、中、高(10、100、500 mg/kg)3個水平濃度,每個水平重復測定6次(實驗結果見表6)。18種PCBs在3個不同加標水平下的回收率為83.9%~104.3%,相對標準偏差RSD為0.3%~4.1%,表明該方法滿足分析要求,適用于茶葉中18種PCBs的定量分析。
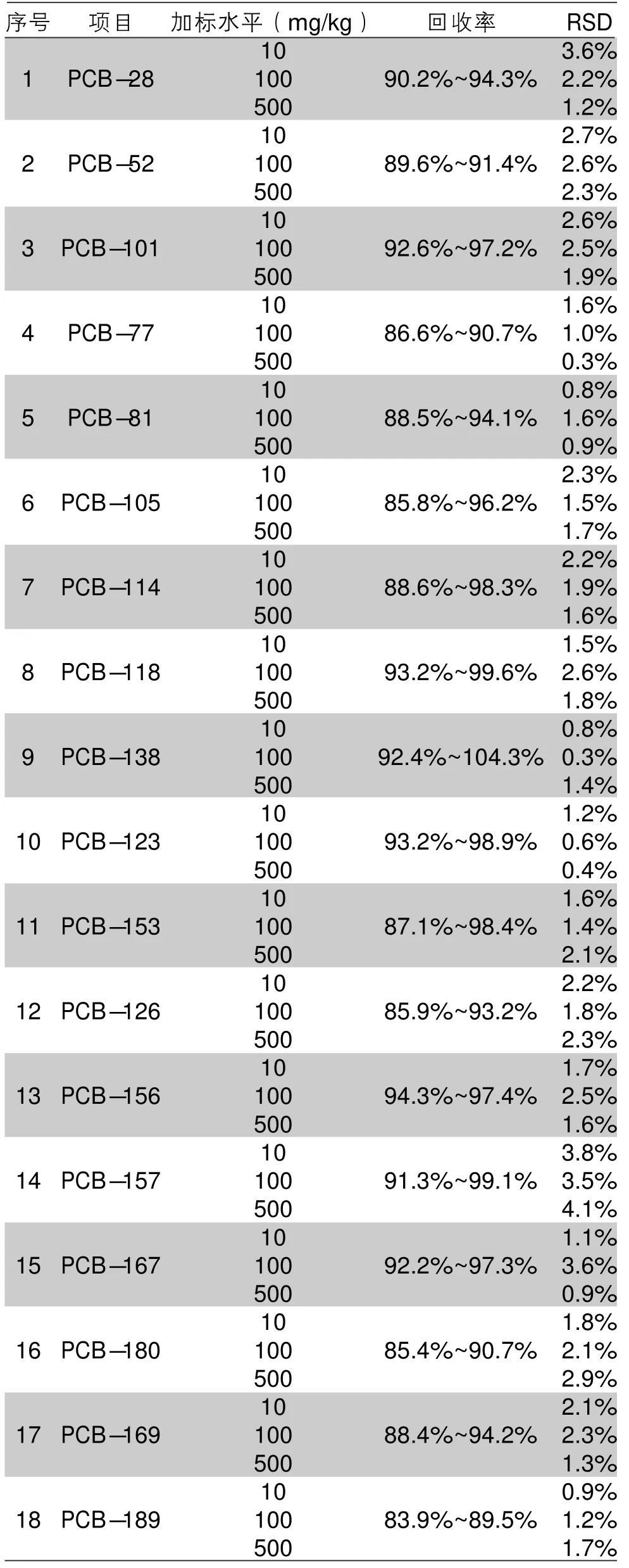
表6 鐵觀音在不同加標水平下18種PCBs的回收率和精密度
2.4 實際樣品檢測
本研究選取網(wǎng)售及市售的38個茶葉樣品,按照前述優(yōu)化后的方法對18種PCBs進行的測定,所有樣品均未檢出(低于檢測限),表明鐵觀音茶葉樣品中多氯聯(lián)殘留的風險相對較低。
3 結論
本研究通過優(yōu)化氣相色譜、質譜等測試參數(shù)建立一種茶葉中18種PCBs的GC-MS/MS法。與傳統(tǒng)的氣相色譜法、氣相色譜質譜法相比,該方法具有靈敏度高、定量準確、操作簡便、分析成本低等特點,能滿足檢驗部門對茶葉中18種PCBs殘留量的快速測定需求,對增強我省茶葉綜合競爭力以及方法標準創(chuàng)新工作具有十分重要的意義。
