牛病毒性腹瀉/黏膜病病毒熒光定量PCR檢測方法的建立
2022-01-04 07:20:40郭志廷仇正英張景艷王貴波梁芬芬李建喜
福建農業學報 2021年9期
張 康,郭志廷,仇正英,張景艷,王 磊,張 凱,王貴波,梁芬芬,馬 倩*,李建喜*
(1.中國農業科學院蘭州畜牧與獸藥研究所/農業農村部獸用藥物創新重點實驗室/甘肅省新獸藥工程重點實驗室,甘肅 蘭州730050;2.甘肅農業大學動物醫學院,甘肅 蘭州730050)
0 引言
【研究意義】牛病毒性腹瀉病毒(bovine viral diarrhea virus,BVDV)隸屬黃病毒科、瘟病毒屬,是一個較大的ORF(開放閱讀框)[1?2],主要包括三個部分3′UTR(3′非翻譯區)、ORF編碼區、5′UTR(5′非翻譯區)。5′UTR序列在整個基因系列中具有高度的保守性,根據這組基因的特點設計引物能夠在分子水平對BVDV進行診斷,并對其進行病原基因分型[3]。目前BVDV是一種威脅全球養牛業的重要病原體[4]。BVDV能夠穿過懷孕母牛的胎盤屏障感染胎兒,從而生出攜帶有大量病原的持續感染(PI)犢牛[5]。該PI牛通常出生后體弱多病,并且由于其自身免疫系統的損傷,特別容易伴隨機會性感染。PI牛在臨床上通常出現死亡率極高的遲發型粘膜病[6],嚴重影響牛群健康,對養殖業造成極大的經濟損失[7]。因此控制BVDV感染最有效的手段就是PI牛的早期診斷[8]。【前人研究進展】目前國內外檢測BVDV的方法繁多,包括免疫組織化學(IHC)、抗原ELISA、RT-PCR和RT-qPCR以及最近開發的RT-ELISA和交叉引物恒溫擴增技術[9]。Driskell等[10]建立檢測BVDV的免疫組織化學(IHC)方法,其優點是適合批量臨床樣本檢測,但需要操作熟練的穩定人員和新鮮完整的組織樣本;李家偉等[11]建立牛病毒性腹瀉病毒LAMP檢測方法,其優點是可現場操作,但極易出現假陽性;范晴等[12]與胡俊英等[13]建立的牛病毒性腹瀉病毒抗原ELISA檢測方法,其包被蛋白暫不能批量生產滿足臨床檢測需求;王榮等[14]提出的使用實時熒光定量RT-PCR檢測BVDV的方法不如探針技術敏感性高。而我國診斷BVDV國家標準技術規定首先采用培養細胞分離病毒的方法,其分離鑒定法主要通過接種牛腎細胞,一般需傳至1~3代左右才能出現明顯病變,具備分離條件的實驗少,因此不適用批量檢測臨床樣本[15]。【本研究切入點】以上每個檢測方法在靈敏度、成本和耗時方面都存在著優缺點。而熒光定量PCR(real-time PCR)技術是一種能夠實時、快速、敏感、準確地檢測病毒在體內的分布及變化且能克服其他方法檢測不足的技術,因此在病原診斷、病原定量檢測及抗病毒機制等方面得到了廣泛應用[9,16]。應用熒光定量PCR方法診斷牛病毒性腹瀉病毒的研究尚待深入探討。【擬解決的關鍵問題】本研究對GenBank提交的BVDV 5′UTR基因序列進行比對,對TaqMan通用探針、BVDV的引物進行改造、設計,以期創建具有靈敏度高、特異性強、能夠相對定量且快速便捷的檢查方法,為獸醫工作者研究和快速診斷牛病毒性腹瀉病毒提供技術支持。
1 材料與方法
1.1 病毒和樣品
從中國獸醫藥品監察所購進BVDV陽性樣品,編號為CVCC AV67,存放于中國農業科學院蘭州畜牧與獸藥研究所獸醫室實驗室。牛傳染性鼻氣管炎病毒(IBRV)疫苗購自金宇保靈生物藥品有限公司,口蹄疫病毒(FMDV)滅活疫苗購自中農威特生物科技股份有限公司,豬瘟病毒(CSFV)疫苗、豬繁殖與呼吸障礙綜合征病毒(PRRSV)疫苗購自青島易邦生物工程有限公司。從甘肅、青海和陜西等省份的奶牛養殖戶中收集到65份臨床癥狀疑似受牛病毒性腹瀉病毒感染的腹瀉牛糞便樣品(獸醫根據腹瀉牛體格瘦小、抵抗能力差、免疫力低下和使用抗生素治療無效等篩選出疑似BVDV感染牛進行采樣)。
1.2 主要儀器及試劑
熒光定量PCR儀,紫外分光光度計,凝膠成像儀;Dnase-FreeRNase,Rnas-Free DnaseI,T7 RNA Polymerase,MaximaH M inus First StrandcDNA Synthesis Kit;2×EsTaqMasterM ix;病 毒DNA/RNA提取試劑盒、DNA瓊脂糖膠回收試劑盒、EasyPURE RNA Purification Kit、One Step PrimeScript TM RT-PCR Kit購自寶生物工程(大連)有限公司。
1.3 引物設計和合成
引物的設計登錄NCBI從GenBank下載BVDV 5′UTR保守區域基因序列,使用MegA lign軟件對其進行對比,選擇其相對保守位置,利用Oligo6.0進行分析,設計2對特異性引物(標準品引物A-F/BR和定量引物Q-F/Q-R)和1條探針,其中標準品上已加有T7啟動子,下游引物加有多聚T尾巴(表1),均由寶生物工程(大連)有限公司合成。
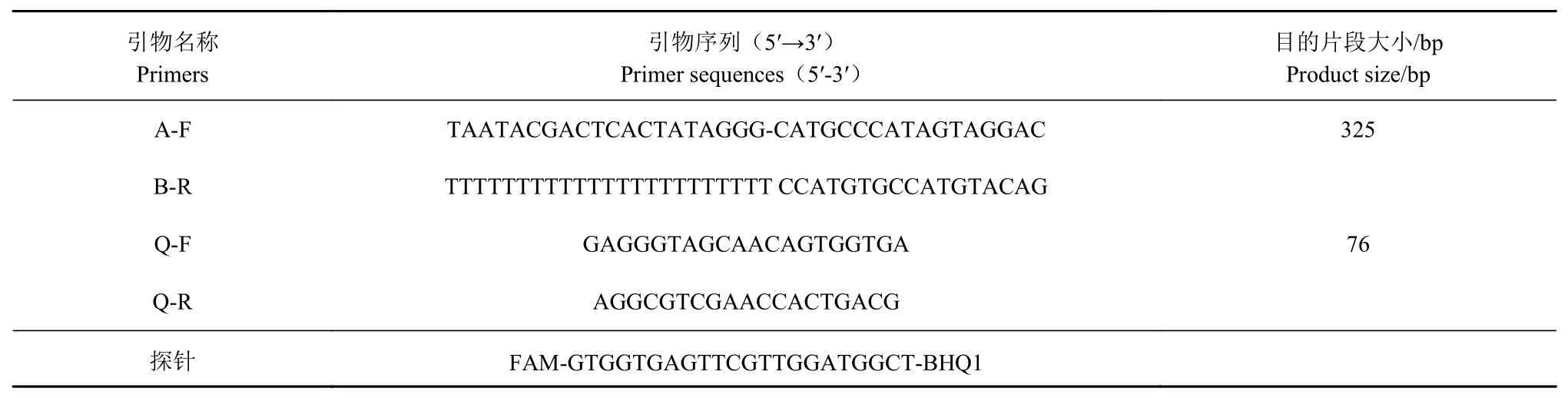
表1 引物和探針Table 1 Prim ersand probesapp lied
1.4 樣品RNA的提取
按照試劑盒中的說明書進行操作,樣本中提取總RNA,置于-20 ℃環境保存備用。
1.5 陽性標準品的制備
以陽性樣品提取的RNA 作為模板進行RNA反轉錄,以引物A-F和B-R作為擴增引物進行PCR產物的擴增,依照膠回收試劑盒的說明對其進行操作,可以得到純化回收目的片段,之后按照T7RNA Polymerase相關說明,對其轉錄進行操作,轉錄后純化所得到的產物即為體外轉錄所制備的標準品。
1.6 熒光定量PCR方法的建立
以BVDV的陽性標準品作為模板建立熒光PCR方法,配制的熒光PCR反應體系包括以下組分:2×One Step RT-PCR Buffer III反應液10μL,TaKaRa ExTaqHS反應液0.4μL,PrimeScript RT Enzyme M ix II反應液0.4μL,上游引物(10μmol·L?1)和下游引物(10μmol·L?1)各0.4μL,熒光探針(5μmol·L?1)0.8μL,RNA標準品2μL,用ddH2O補充至20μL,擴增條件為:溫度45℃時間5m in,95℃10 s,40個擴增循環(95℃5 s、60℃20 s)。
1.7 標準擴增曲線的建立和敏感性試驗
將標準品RNA模板濃度分別進行10倍倍比稀釋,以5.0267×109copies·μL?1為 起 始 濃 度 稀 釋 到5.0267×10?1copies·μL?1,之后按照其相關要求對其進行稀釋,不同稀釋度核酸作為模板進行PCR擴增,將相關標準濃度的Ct值作為縱軸指標,以稀釋倍數所計算得到的對數作為橫軸指標,畫出其標準曲線。在這個曲線基礎上得到熒光定量PCR的敏感程度以及最小測定值。
1.8 特異性試驗
按照上述的手段對其FMDV、BVDV、IBRV、CSFV、PRRV的DNA/RNA模板進行擴增,對其定量PCR反應指標進行觀察,對特異性進行評價。
1.9 重復性試驗
將得到的已知濃度BVDV陽性標準品按照10?2、10?3、10?4、10?5以及10?6的比例進行稀釋,然后進行組間與組內重復試驗,在進行組內試驗的過程中,需要將每一個標準使用同一種方法重復3次;在進行組間試驗時,需要在不同的時間使用這一方法重復3次,針對測定的結果對其進行分析,來評估本次研究的價值,能否將結果復現。
1.10 熒光PCR方法的應用
使用已經獲得的熒光定量PCR方法,對臨床收集到的65份標本(這些標本均為懷疑為牛病毒性腹瀉病毒感染的牛糞便)進行擴增,出現了陽性結果并對本次研究的可行性程度進行評估。
2 結果與分析
2.1 BVDV標準品引物的特異性擴增結果
按照相關說明書對其病毒RNA進行擴增操作,產物經1.0%瓊脂糖凝膠電泳,對其進行測定,獲得了分子量為325 bp左右的條帶(圖1)。
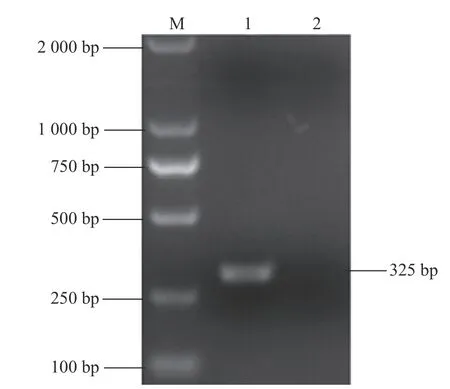
圖1 標準品引物擴增產物Fig.1 Am p lification productsof RNA standard primers
2.2 標準品RNA純度分析結果
紫外分光光度計測定結果顯示,標準RNA原液的D260/D280值達到1.92,其質量濃度達到35.70mg·L?1。
2.3 熒光定量PCR方法的建立和特異性試驗
將BVDV陽性核酸作為模板,對其進行擴增,最終得到了S形曲線,同時用建立的熒光探針方法對FMDV、IBRV、CSFV和PRRS病毒進行擴增,上述樣品都沒有得出特異性條帶,這就證實這一檢測手段對BVDV有較好的特異性,但對遺傳物質比較類似的病毒并沒有出現相關的反應(圖2)。
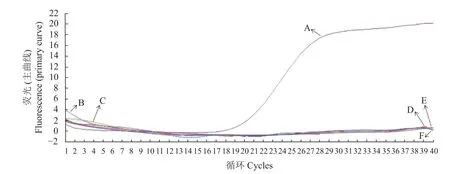
圖2 熒光定量PCR方法特異性試驗Fig.2 Specificity of RT-PCR assay
2.4 熒光定量PCR方法的標準曲線和敏感性試驗
將BVDV陽 性 核 酸(含 量 為5.026 7×109copies·μL?1)10倍稀釋后進行擴增(表2),其數據表明,由于模板拷貝數不斷減少,Ct值逐漸上升,當稀釋達到10?1、10?2、10?3、10?4、10?5、10?6、10?7、10?8和10?9時,Ct值 分 別 為11.17、13.48、18.68、21.43、24.13、28.44、31.22、35.16和37.69,為S形曲線,但達到10?10時,則不顯示曲線。這表明本方法的檢測極限是樣品的10?9稀釋倍數,即陽性標準品的濃度為5.026 7 copies·μL?1(圖3)。以10?2、10?3、10?4、10?5、10?6、10?7和10?8等不同濃度的反對數作為橫軸,以對應的Ct值作為縱軸制作標準曲線(圖4),可以得到相關的回歸方程:y=3.468 9x+7.303 9,相關系數R2=0.993 8,這說明Ct值與拷貝數之間呈現非常強的線性關系,表明該方法穩定性好,意味著無論待檢樣品的模板濃度高低,用該方法進行檢測都可以得到較好的試驗結果。
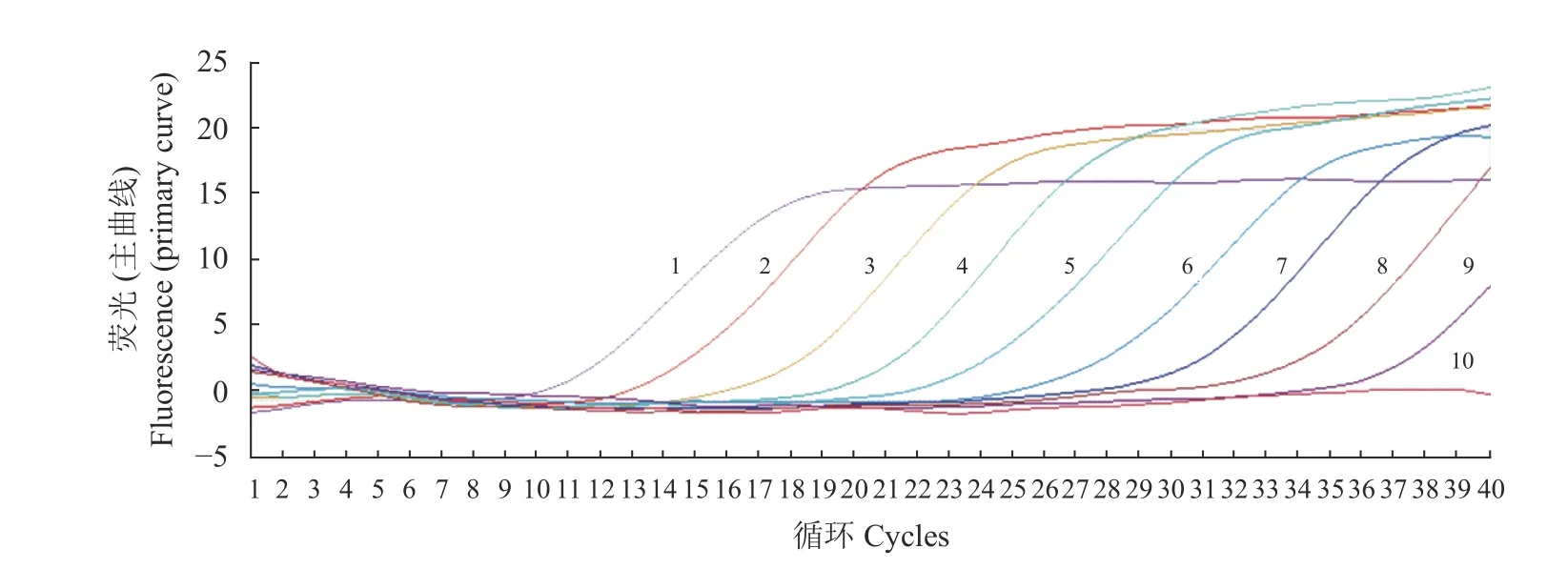
圖3 熒光定量PCR敏感性試驗Fig.3 Sensitivity of RT-PCR assay
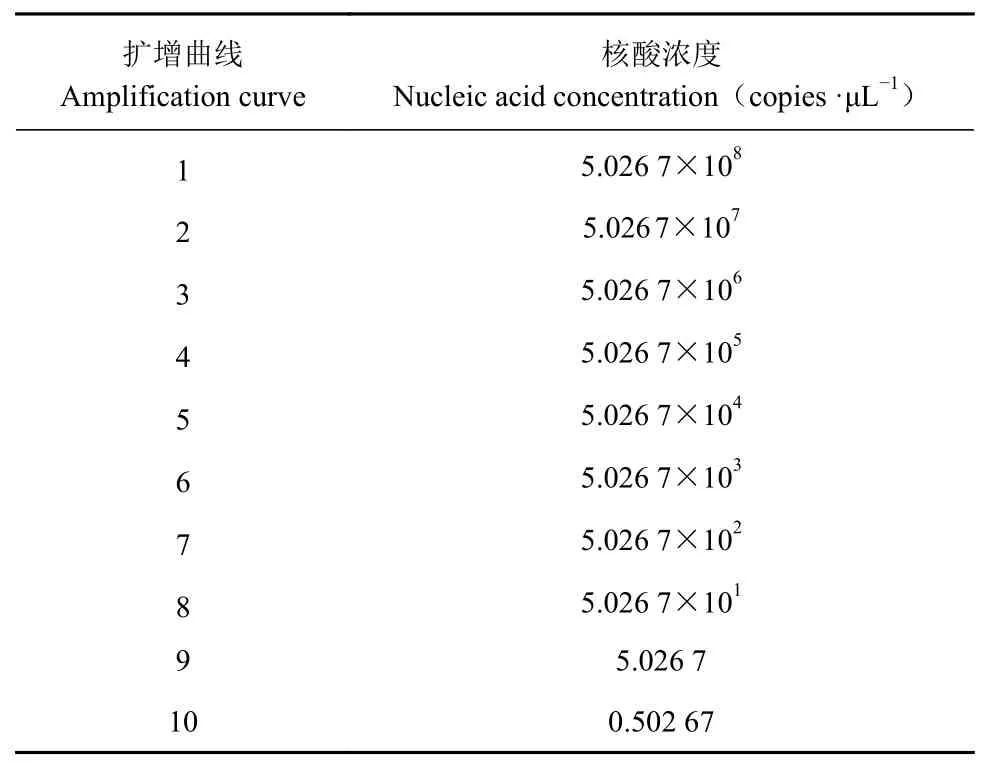
表2核酸濃度Tab le 2 Nucleic acid concentration
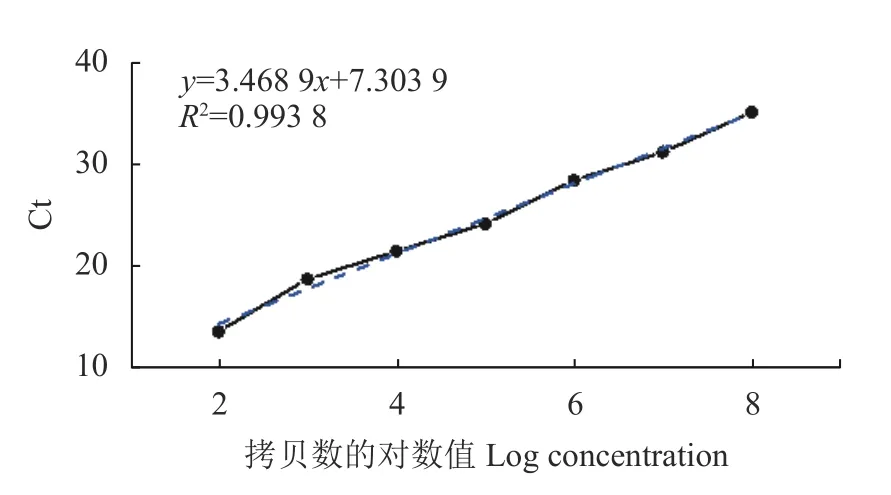
圖4 熒光定量PCR標準曲線Fig.4 Standard curve for RT-PCR assay
2.5 熒光PCR方法的重復性
獲取以 上的稀釋 倍數10?4、10?5、10?6、10?7和10?8的模板對其進行重復性研究,結果表明,組內重復試驗的變異系數(CV)平均為0.77%±0.13%,組間重復試驗的變異系數(CV)平均為0.90%±0.67%,這表明本方法的重復性較好,結果詳見表3和圖5。

圖5 熒光定量PCR重復性Fig.5 Reproducibility of RT-PCR assay
2.6 熒光定量PCR方法的應用
用建立的熒光PCR方法對65份腹瀉牛肛門拭子樣品進行檢測,共有4份樣品得到了S型曲線(圖6),與陽性數據相符,說明這一方式是可行的,在實際中可以使用到該疾病的流行病學監測以及篩查PI牛等方面。
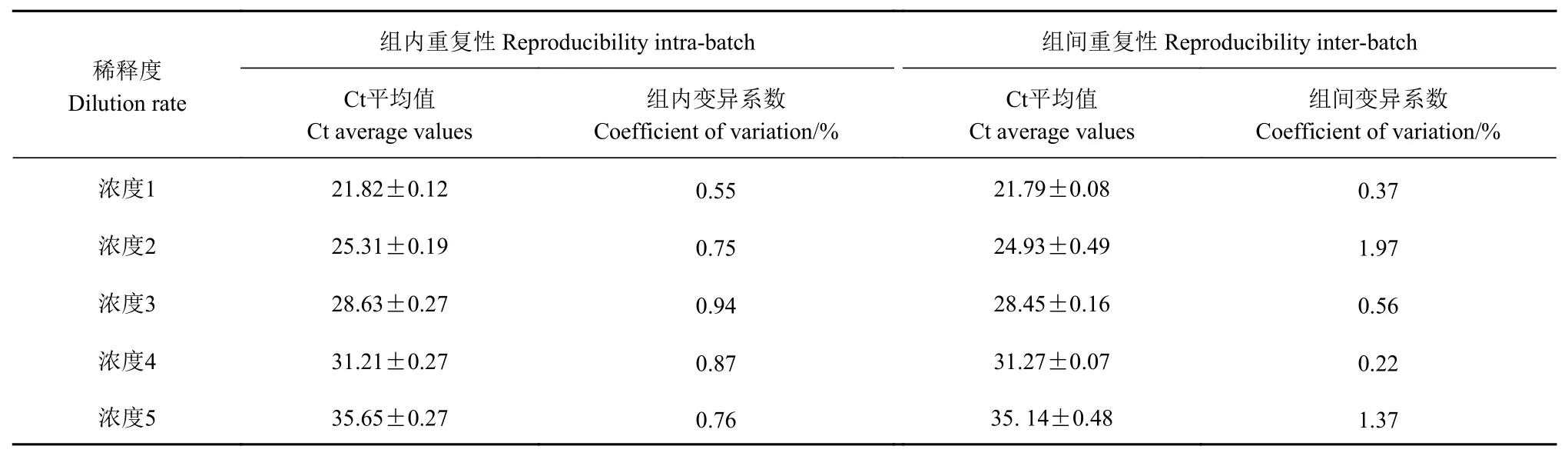
表3實時熒光定量PCR方法的變異系數Table3 Coefficientsof variation between inter- and intra-groupsof RT-PCR assay
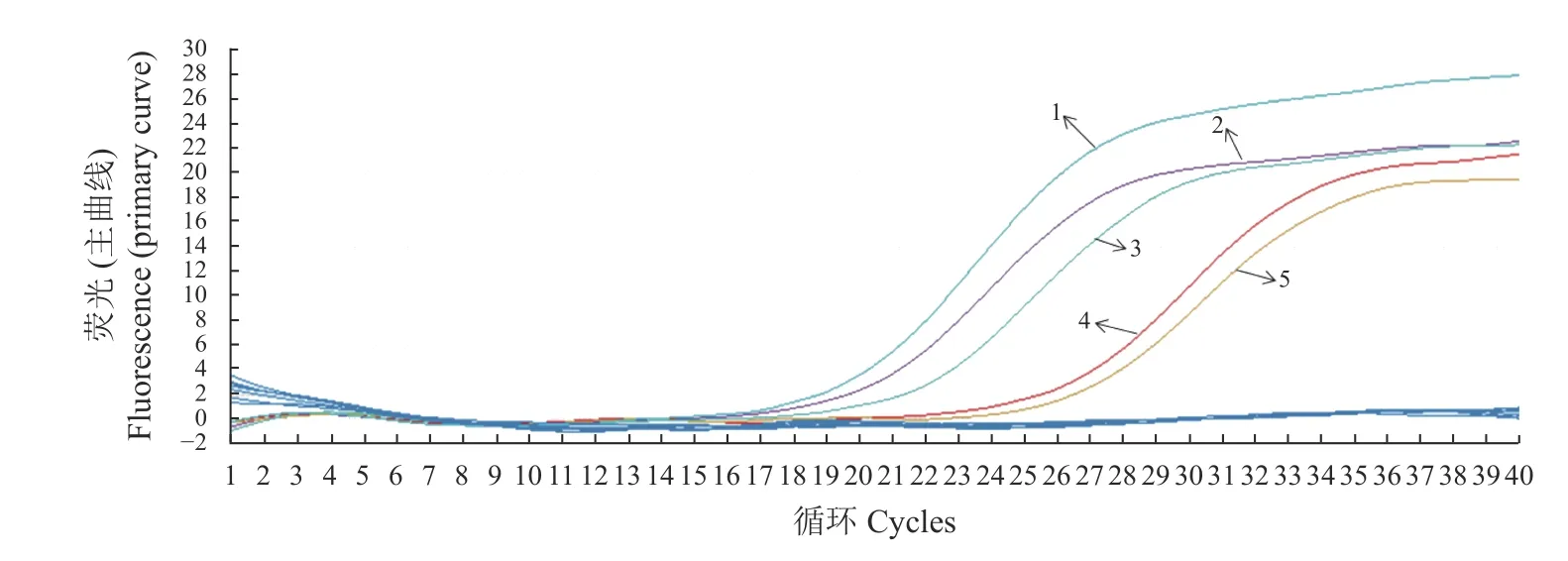
圖6 熒光定量PCR方法的應用Fig.6 App lication of RT-PCR assay
3 討論與結論
早期篩查PI牛是控制該病毒傳播的重要措施之一。ELISA檢測技術一直被廣泛用于檢測血液、血清、乳汁、鼻腔和口腔棉拭子中BVDV抗體水平,根據抗體水平判定是否為PI牛,而ELISA往往由于母源抗體的影響造成檢測失敗[17]。因此近年來國內外研究人員越來越多地利用熒光定量PCR技術檢測BVDV,不僅因為其靈敏度高,最重要的原因是能夠在早期診斷鑒定PI牛,為此本試驗以GenBank中提交的BVDV 5′UTR基因序列進行比對,設計相關的PCR檢測方法。
通過特異性、重復性以及敏感性試驗,本試驗建立的BVDV TaqMan實時熒光定量RT-PCR檢測方法,對FMDV、BVDV、IBRV和PRRS等病毒無特異性擴增。該方法靈敏度高,對病毒檢測的最低檢測靈敏度可達到5.026 7 copies·μL?1,敏感性優于常規的RT-PCR[18];穩定性好,標準曲線相關系數是0.99 6;重復性較好,同一時期組內和不同時期組間變異系數均小于1.0%。
本試驗建立的熒光定量PCR方法與常規PCR檢測方法相比不需在反轉錄后再次向反應混合物中添加任何試劑,僅在一支PCR反應管中操作,直接由RNA定量RNA,提高了操作效率,而且減少了開管次數從而有效降低外界其他基因污染的風險[19?20]。
本試驗顯示該方法專一測定這一病毒,因此該方法既可以用于臨床早期發現PI牛,又可以檢測細胞中BVDV的增殖情況,這為研究細胞水平上藥物抗BVDV及其分子機制提供了檢測手段。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
當代陜西(2019年8期)2019-05-09 02:22:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
專用汽車(2016年4期)2016-03-01 04:13:43
Coco薇(2015年1期)2015-08-13 02:47:34
