第一性原理研究3C-SiC/Mg復合材料界面的電子結構
2022-03-04 02:32:54堯軍平唐錦旗陳致君
原子與分子物理學報 2022年4期
呂 昭,堯軍平,唐錦旗,陳致君
( 南昌航空大學 航空制造工程學院,南昌 330063)
1 引 言
鎂合金有著導電導熱性能良好、剛性好、密度低等特點,關于鎂合金的應用、研發備受各界關注,但鎂合金由于耐蝕性能差、強度低、易燃性等缺點使得鎂合金不能滿足人類在更多場合實際應用的需要,由此對鎂合金新型材料的研發制備迫在眉睫.鎂基復合材料擁有高比強度、高比鋼度、高比模量等特性,被認為是21 世紀高新技術產業中最有希望被大量利用的輕金屬材料之一,廣泛的應用在汽車制造、電子設備、航空航天、國防裝備等領域[1-4].
界面的結合性能對于復合材料有著重要的影響[5],DFT( Density functional theory) 在界面性能的研究方面有突出貢獻,能較為直觀的給出界面處原子位置,而且可以在電子級別分析界面的結合機制[6,7].而近年來計算機技術的飛速發展,計算機模擬技術逐漸在材料的開發研究中變得重要起來,材料的研發模式開始由傳統的實驗方式逐步轉為理論模擬.其中第一性原理的計算方法又是其中最為可靠的.例如,Shingo 等人[8]對3C-SiC(111) /Al 界面進行了第一性原理贗勢計算,3C-SiC(111) 的C 終端界面的結合能大于Si 終端界面的結合能.Liu R 等人[9]研究了Mg( 0001) /TiB2(0001) 界面的界面結構,發現Mg 原子與Ti端TiB2表面和B 端TiB2表面結合時,分別形成金屬/共價混合鍵和離子鍵; Mg/TiB2界面的界面能遠大于α - Mg/Mg 界面.Wu 等人[10]研究了Al(111) /6H-SiC(0001) 的界面結合及沿Z 軸拉伸時的界面斷裂特性,結果表明,在Al( 111) /6H-SiC(0001) 界面,C 端接界面的粘附功為2.689 J/m2,高于端接界面的1.649 J/m2.在抗拉強度方面,C 端接界面高于Si 端接界面.兩種復合材料的強度均高于純鋁,但延展性較弱.熊輝輝等人[11]用第一性原理方法計算了6 種不同Ti(0001) /TiB2( 0001) 界面的黏附功和界面能,研究結果表明,在6 種不同的界面中,B 終端的Ti/TiB2界面穩定性均優于Ti 終端的界面,且B 終端的孔穴位堆垛界面( BTH) 和Ti 終端的心位堆垛界面( TTC) 分別是兩種終端最穩定的界面.
陶瓷顆粒增強金屬基復合材料界面結合性能研究目前是國內外學者研究熱點,但對于SiC/Mg界面結合性能研究未見報道.基于密度泛函理論的第一性原理對于界面特性研究有著優勢,可以從微觀方面研究作用機理和界面特性[10,12-15],可以滿足對SiC/Mg 金屬-陶瓷復合材料界面結合機制的研究.本文通過第一性原理研究了4 種搭建結構的3C-SiC(111) /Mg(0001) 界面模型,通過分析理想粘附功和電子結構得出穩定的結構.
2 計算方法及模型
2.1 計算步驟和細節
本文所有的計算都在MATERIALS STUDIO 軟件的CASTEP 模塊下進行[16,17],描述電子與電子之間作用的交換關聯勢泛函的選取為GGA-PBE,優化算法選取BFGS,贗勢采用平面波超軟贗勢.能量的計算采用自洽迭代方法( SCF) ,自洽迭代的收斂閾值為1.0 ×10-6eV /原子,能量的截斷點為380 eV,能量計算允許的最大自洽迭代次數為100,幾何優化時作用在原子上的最大力應不超過0.03 eV/?,最大應力應不超過0.05 GPa,最大位移應不超過0.001 ?,第一布里淵區K 值為6 ×6 ×1.
Mg 原胞由兩個Mg 原子構成,空間群為P63/mmc( No.194) ,晶格參數為a=b=3.2094 ?,c=5.2105 ?,α=β=90°,γ=120°.3C-SiC 又名β-SiC,具有閃鋅礦結構,空間群為F-43m,它的原胞由4 個Si 原子和4 個C 原子組成,四個相近的原子將中間的異種原子包圍而形成一個正四面體.晶格參數a=4.348 ?.
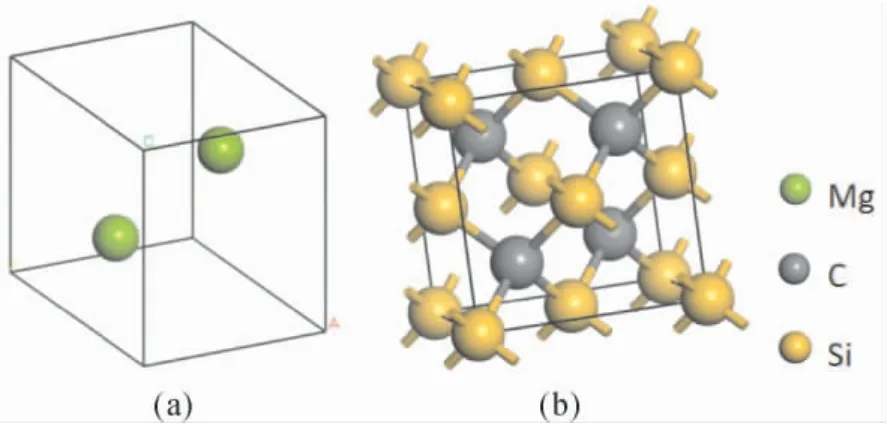
圖1 ( a) Mg 的晶體結構; ( b)3C-SiC 的晶體結構Fig.1 ( a) Crystal structure of Mg; ( b) Crystal structure of 3C-SiC
2.2 模型建立
圖2 顯示的是3C -SiC(111) /Mg(0001) 的界面模型,通過在10 層的3C -SiC( 111) 上堆疊5層Mg(0001) 來實現,在Mg(0001) 側的上方建立15 ? 厚度的真空層,防止表面原子之間的相互作用.對于3C-SiC(111) 是極性表面,有著兩種封端結構,因此采用兩種封端來模擬3C - SiC(111) /Mg( 0001) 的界面.本文還考慮了Mg(0001) 面和3C -SiC( 111) 面可以構建中心型和頂位型兩種堆垛結構,總共建立4 個模型用于接下來的計算分析.
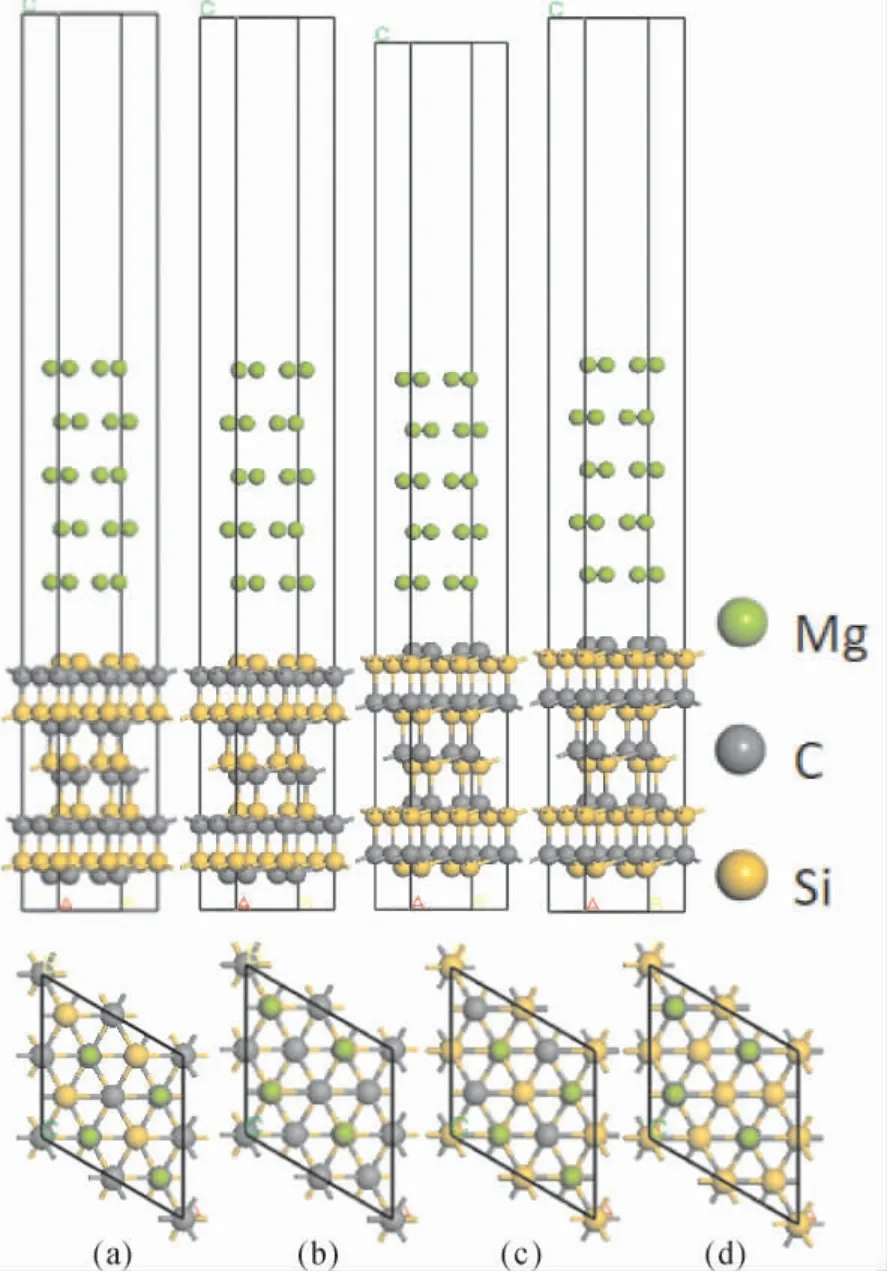
圖2 3C-SiC(111) /Mg( 0001) Si 終端和C 終端的四種結構: ( a) Si 終端中心型; ( b) Si 終端頂位型; ( c) C 終端中心型; ( d) C 終端頂位型Fig.2 Four structures of Si-terminated and C-terminated 3C-SiC(111) /Mg(0001) : ( a) Center-site Si-terminated; ( b) Top -site Si -terminated; ( c) Center - site C - terminated;( d) Top-site C-terminated
3 結果與討論
3.1 結構優化
結構優化計算過后,3C - SiC ( 111) /Mg(0001) 的4 種界面模型在z 軸方向原子都發生了位移,使得模型的界面間距產生了不同程度的縮小,達到了每個界面較穩定的狀態.
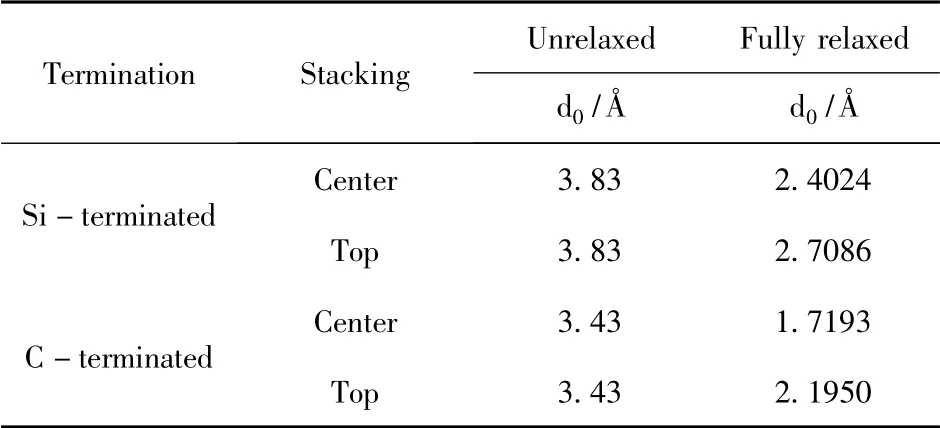
表1 3C-SiC(111) /Mg(0001) 界面結構優化前后的界面間距Table 1 Interface spacings before and after structure optimization of 3C-SiC(111) /Mg(0001)
3.2 理想粘附功
金屬/陶瓷界面結合強度的表示通常用分離功這個物理量,分離功的對立是粘附功,粘附功的含義是指兩個自由表面結合形成一個界面所釋放的能量,其計算公式如下[18]:
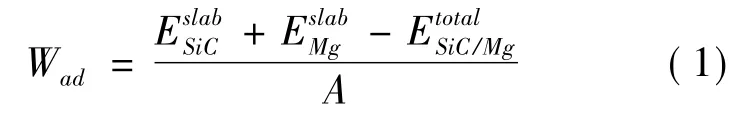
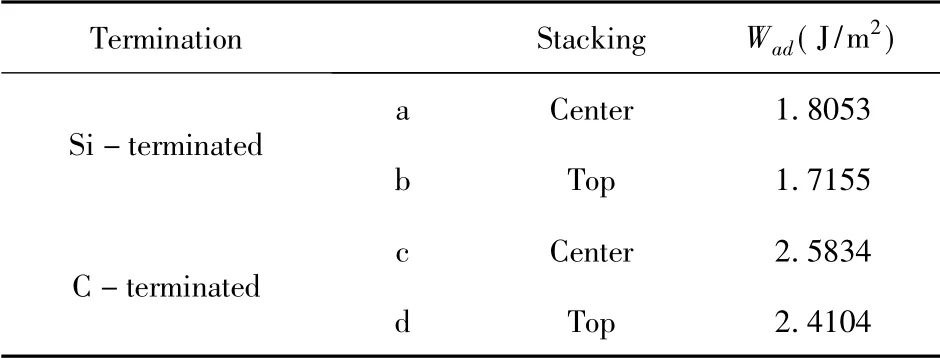
表2 3C-SiC(111) /Mg(0001) 界面的粘附功Table 2 Adhesion work of 3C - SiC( 111) /Mg( 0001)interface
結構優化之后,4 個模型在z 軸方向都發生了界面距離不同程度的減少,界面間距與界面結合強度有著緊密的關系,而在4 個3C - SiC(111) /Mg(0001) 界面模型中,C 封端模型的界面間距比Si 封端模型更小,粘附功而言,C 封端模型的更大,相對來說,C 封端模型明顯更穩定.該結果與以往的研究[19-21],當SiC 與金屬結合時,用C 封端與金屬面接觸往往表現出更強的穩定性相一致.而在Si 封端和C 封端的模型中,各自的中心型模型都優于頂位型模型,有著更小的界面間距(2.4024 ? 和1.7193 ?) 和更大的粘附功(1.8053 J/m2和2.5834 J/m2).
3.3 電子結構分析
圖3( a) 和圖3 ( b) 分別是3C -SiC(111) /Mg(0001) Si 終端中心型結構的電荷密度分布和電荷密度差,圖3( c) 和圖3 ( d) 分別是3C - SiC(111) /Mg(0001) C 終端中心型結構的電荷密度分布和電荷密度差.圖3( a) 和圖3 ( c) 中Mg 側和SiC 側中間有電荷積累.SiC 側的電荷密度主要聚集在C 原子附近,這是因為C 原子相對于Si 原子有較強的電負性.圖3( b) 和圖3( d) 可以看出,Si 原子附近主要是代表著失去電子的藍色區域,C 原子附近主要是代表得到電子的紅色區域.在界面處存在電子云重疊,圖3( d) 的電子云重疊現象明顯于圖3( b).界面處的Mg 原子失去的電子進入界面,與界面處的Si 原子和C 原子的電荷形成離子鍵/共價鍵.圖3( d) 在界面處積聚電子的現象比圖3( b) 更顯著,表明C 端結構比Si 端結構的界面結合更穩定.這個結論與粘附功的分析結果相一致.
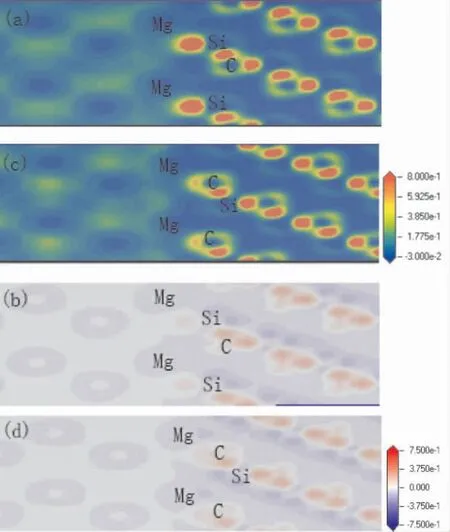
圖3 中心型電荷密度分布圖和差分電荷密度圖:( a) Si 終端電荷密度分布圖; ( b) Si 終端差分電荷密度圖; ( c) C 終端電荷密度分布圖;( d) C 終端差分電荷密度圖Fig.3 Charge density distributions and charge density differences of center -site: ( a) charge density distribution of Si - terminated; ( b) charge density difference of Si - terminated; ( c)charge density distribution of Si - terminated;( d) charge density difference of Si-terminated
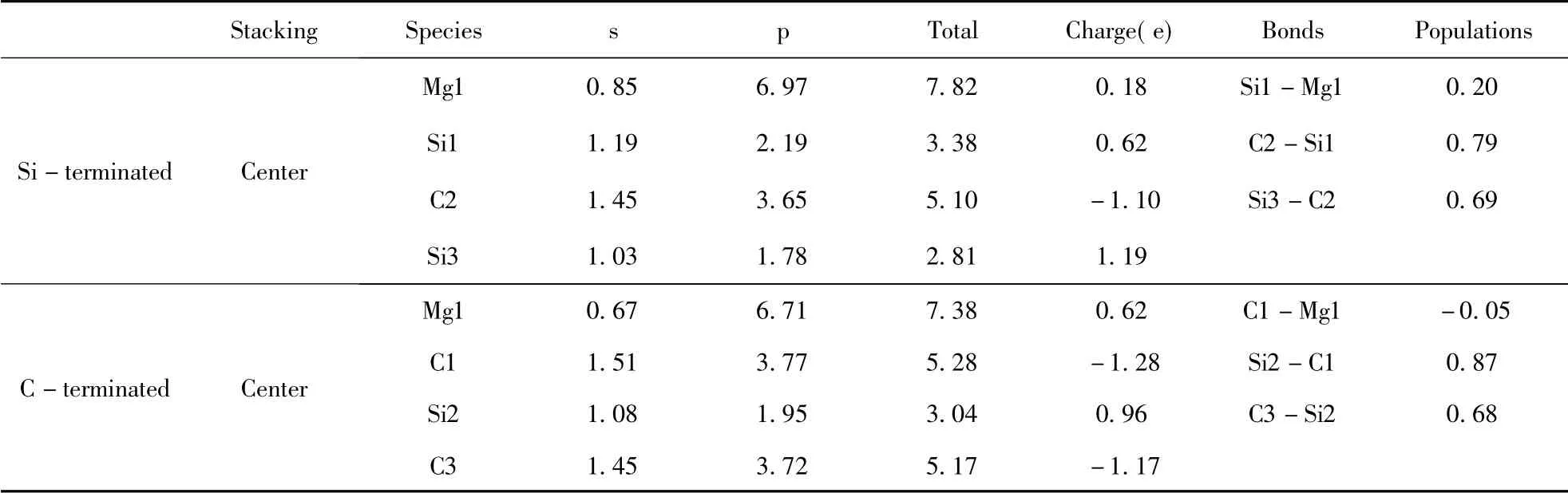
表3 中心型兩種終端結構界面原子的Mulliken 電荷Table 3 Mulliken charges of atoms at center-site
布居數可以反應出電子在界面處的散布狀況以及衡量成鍵類型和強弱,鍵布居數越靠近0 代表離子性越強,越靠近1 代表共價性越強.在Si端3C -SiC( 111) /Mg( 0001) 界面上,界面的Mg原子失去部分電子變成+0.18 價,與界面上的Si原子的重疊布居數為0.20,處于鍵合狀態.與Si端相比,C 端3C -SiC(111) /Mg(0001) 界面上的Mg 原子失去的電子( +0.62) 更多,與界面上的C原子成弱反鍵狀態,重疊布居數為-0.05.Si 端Mg 原子與相鄰原子形成的Si -Mg 鍵的布居數大于C 端Mg 原子與相鄰原子形成的C -Mg 鍵,Si-Mg 鍵的離子性弱于C -Mg 鍵.從表中可以比較得出,Si 端與C 端3C-SiC(111) /Mg(0001) 界面處的Si-C 鍵的重疊布居數(0.79 和0.87) 大于內部的Si-C 鍵的重疊布居數(0.69 和0.68) ,表明界面改變了Si-C 的共價鍵性質.
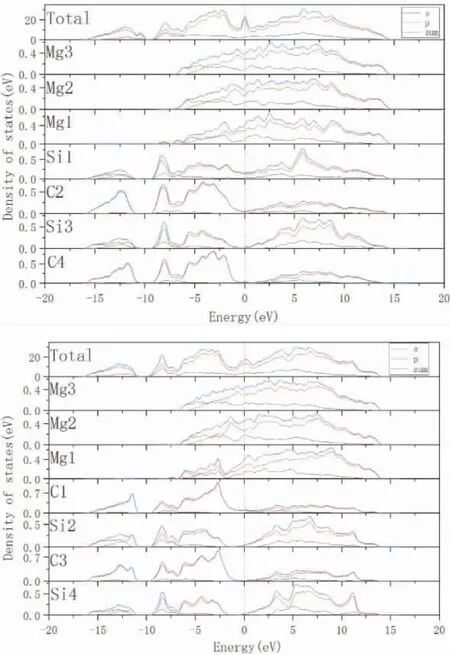
圖4 3C-SiC(111) /Mg(0001) 界面態密度圖: ( a)Si 終端中心型結構; ( b) C 終端中心型結構Fig.4 Density of states of 3C -SiC( 111) /Mg( 0001)interface: ( a) Center - site Si - terminated;( b) Center-site C-terminated
從態密度的計算結果可以看出界面處原子的成鍵性質,離界面比較遠的原子對界面的影響很小,因此計算了3C-SiC(111) /Mg(0001) 中心型結構兩種終端( C 終端和Si 終端) 的界面部分原子的態密度.在圖5( a) 中,Mg 側界面Mg 原子的態密度曲線不同于內部的Mg 原子的態密度曲線,Si側原子的態密度曲線也出現不同.計算結果顯示,Si 側界面Si 原子的態密度曲線高度低于內部C 原子的態密度曲線高度( 從-15.9 eV 到-10.1 eV) ,表明SiC 側界面Si 原子的電子發生轉移到了Mg側的界面Mg 原子,從而形成了離子鍵.由于Sisp、C-sp、Mg-sp 在-9.5 eV 到14.6 eV 之間的軌道雜化,表明有共價鍵的存在.在費米能級附近,Mg 原子和Si 原子的明顯的占據態表明有金屬鍵產生.C 終端原子態密度的分析和Si 終端原子態密度的分析相近,Si 終端和C 終端界面原子成鍵類型主要是共價鍵、離子鍵和少量金屬鍵.
4 結 論
本文采用第一性原理的方法計算了3C -SiC(111) /Mg(0001) 界面的粘附功和電子結構,考慮了界面的中心型和頂位型兩種堆積結構和Si 終端和C 終端兩種端接結構.計算后可以得出以下結論:
(1) 結構優化后,除了界面間距均有或多或少的減少外,這4 種界面模型的結構沒有發生特別大的變化.
(2) 中心型結構的粘附功大于頂位結構,C終端結構的粘附功大于Si 終端結構,C 終端中心型的結構是四個結構中最穩定的.
(3) 根據電荷密度分布圖、差分電荷密度圖、Mulliken 電荷和態密度圖分析,這兩種界面處存在離子鍵、共價鍵和少量金屬鍵的混合.
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
哲學評論(2021年2期)2021-08-22 01:53:34
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
當代陜西(2020年13期)2020-08-24 08:22:02
數學物理學報(2020年2期)2020-06-02 11:29:24
中華詩詞(2019年7期)2019-11-25 01:43:04
制造技術與機床(2017年5期)2018-01-19 02:49:17
濰坊學院學報(2016年2期)2016-12-01 13:00:11
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
光學精密工程(2016年6期)2016-11-07 09:07:19
