基于UPLC-Q/Orbitrap HRMS多目標快速篩查魚肉中30種蛋白同化激素及糖皮質激素
2022-03-05 08:54:56郭添榮吳文林萬渝平陳代偉張龍翼
食品科學 2022年4期
郭添榮,吳文林,,*,張 崟,萬渝平,葉 梅,陳代偉,黃 霞,張龍翼
(1.成都市食品檢驗研究院,四川 成都 611130;2.中國科學院成都生物研究所,四川 成都 610041;3.中國科學院大學,北京 100049;4.成都大學 肉類加工四川省重點實驗室,四川 成都 610106)
激素是由生物體分泌且具有調節或影響機體生長代謝的微量活性物質,主要包括蛋白同化激素、糖皮質激素等[1]。蛋白同化激素具有蛋白質同化作用,可以有效促進骨骼發育壯大,縮短動物生長周期[2-3]。糖皮質激素具有調節動物機體的脂肪、糖類合成和代謝作用,促進動物生長繁殖,因此存在以提高經濟利益為目的在魚類養殖中違規使用的現象[4-6]。研究表明,長期食用蛋白同化及糖皮質激素含量超標的食品,可能引起腎上腺功能亢進、肥胖以及骨質疏松等疾病[7-10]。我國農業部第235號公告和GB 31650—2019《食品中獸藥最大殘留限量》中對激素藥物限量具有明確規定[11],歐盟與美國相關組織也早已頒布了相關禁限令[12-13]。為滿足監管需要,建立一種多目標快速篩查激素殘留的方法顯得十分必要。
目前蛋白同化激素及糖皮質激素殘留檢測方法主要有免疫分析法[14]、高效液相色譜法[15]、液相色譜-串聯質譜法[16-18]、液相色譜-四極桿飛行時間質譜法[19-20]等。其中免疫分析法檢測周期較長且受活性酶種類限制而逐漸被取代,高效液相色譜法僅能分離幾種特定的物質且難以區分相同或相近物質而應用受限,液相色譜-串聯質譜法雖為主流檢測方法,但受到分析模式和分辨率限制,難以實現對復雜樣品的高通量多目標分析,液相色譜-四極桿飛行時間質譜法具有靈敏度高、適用范圍廣等優勢,但因串級功能不全以及面對同分異構體物質分辨率不夠而定性能力受到限制[21]。超高效液相色譜-四極桿/靜 電場軌道阱高分辨質譜(ultra-high performance liquid chromatography-quadrupole/orbitrap high-resolution mass spectrometry,UPLC-Q/Orbitrap HRMS)法將高選擇性的四極桿與超高分辨率、高靈敏度的靜電場軌道阱有機結合,針對復雜樣品基質的多種激素檢測,在建立激素數據可追溯性電子身份數據庫的基礎上,實現在無標準品對照情況下對化合物進行大批量多目標快速篩查[22-23],目前已被廣泛應用于組學研究、環境檢驗以及違禁藥物檢查等[24-27]眾多領域,研究報道多應用于肉類食品與保健品中多肽類、大環內酯類以及糖皮質類[28-29]的篩查,本實驗將該技術用于魚肉中30種蛋白同化及糖皮質激素的多目標快速篩查研究,以期為魚類中激素殘留風險監測提供有效的技術參考。
1 材料與方法
1.1 材料與試劑
乙腈、甲醇(均為色譜級) 美國Thermo Scientific公司;甲酸、乙酸銨(均為色譜級) 美國Sigma Aldrich公司;Oasis?HLB、Oasis?PRiME HLB固相萃取柱(6 mL 200 mg) 美國Waters公司;Captiva EMR Lipid固相萃取柱(6 mL 600 mg)、陶瓷均質子(100個/瓶) 美國Agilent公司;超純水(電阻率為18.2 MΩ·cm,25 ℃) 美國Millipore公司;10種蛋白同化激素標準品(去氫睪酮、表睪酮、氟甲睪酮、甲睪酮、諾龍、丙酸睪酮、群勃龍、美雄酮、苯丙酸諾龍、美睪酮) 美國Cato Research Chemicals Inc公司;20種糖皮質激素標準品(丙酸倍氯米松、倍他米松、醋酸可的松、氫化可的松、甲基潑尼松龍、潑尼松龍、曲安奈德、曲安西龍、可的松、地塞米松、醋酸氫化可的松、倍氯米松、氟米松、布地奈德、醋酸氟輕松、醋酸氟氫可的松、氟米龍、丙酸氯倍他索、醛固酮、潑尼松,純度均不小于95.0%) 中國食品藥品檢定研究院;36 批次魚(多寶魚、鱖魚、烏魚和草魚各9 批次) 市購。
1.2 儀器與設備
Vanquish Horizen-Q Exactive Plus高分辨質譜系統 美國Thermo Fisher公司;ORTEX3旋渦混勻器、 T18 basic均質機 德國IKA公司;3K15高速冷凍離心機 美國Sigma Aldrich公司;Turbovap LV濃縮氮吹儀 美國Biotage Caliper Zymark公司;Milli-Q超純水系統 美國Millipore公司;ME203型電子天平 瑞士Mettler Toledo公司。
1.3 方法
1.3.1 樣品前處理
準確稱取2.00 g(精確至0.01 g)勻漿試樣,置于50 mL聚丙烯離心管中,加入80%乙腈溶液(含0.2%甲酸)10 mL,靜置10 min后再加入兩粒陶瓷均質子,快速劇烈振搖并旋渦振蕩30 min(2 000 r/min)使樣品充分散開,9 500 r/min高速離心5 min,取上清液待凈化。取5 mL準確量取的上清液過6 mL 200 mg的Oasis PRiME HLB固相萃取柱,流速為1 滴/s,收集4 mL后半段流出液并準確量取2 mL,氮氣下吹至近干,初始流動相定容至1 mL,過0.22 μm有機濾膜,同時做試劑空白實驗一同上機測試。
1.3.2 儀器條件
UPLC條件:Acquity BEH C18色譜柱(2.1 mm× 100 mm,1.7 μm);色譜柱溫度25 ℃;流速0.5 mL/min;流動相A(水相)為含0.1%甲酸的乙酸銨(20 mmol/L) 溶液,流動相B(有機相)為乙腈;梯度洗脫程序:0~0.5 min,95% A、5% B;0.5~15 min,2% A、98% B;15~20 min,2% A、98% B;20~20.1 min,95% A、5% B;20.1~25 min,95% A、5% B。進樣量10 μL。
HRMS條件:加熱電噴霧離子源;掃描采集方式:正負離子同時采集,一級全掃描/數據依賴二級掃描(Full MS/dd-MS2)(Top N);噴霧電壓3.5 kV(+),-3.0 kV(-);離子傳輸管及加熱器溫度325 ℃和450 ℃;鞘氣及輔助氣(N2)流速:35 arb和10 arb;質量掃描范圍m/z100~1 000;分辨率75 000(Full MS)、17 500(dd-MS2);C-Trap中離子數最大容量:3×106(Full MS)、2×105(dd-MS2);C-Trap中最大注入時間:100 ms(Full MS)和50 ms(dd-MS2)。
1.3.3 數據庫的建立
本研究基于Xcalibur 4.1中TraceFinder 4.1和mzVault軟件搭建數據庫。首先收集30種激素化合物的中英文名稱、分子式、CAS號等基本信息,并在Full MS全掃描模式下獲取30種化合物的準確保留時間、母離子精確質量數等信息(表1),隨即導入數據庫中,得到可用于高通量快速篩查的指紋識別數據庫。在dd-MS2模式下,經不同碰撞能量下發生裂解得到的高能量碰撞解離(high energy collision dissociation,HCD)碎片離子質譜圖匯總形成用于進一步譜圖匹配確證的譜圖庫。
1.3.4 基質效應的計算
將30種激素標準品分別通過4種魚肉基質以及甲醇制備為標準溶液,通過甲醇純溶劑和基質溶液中同一物質的標準曲線斜率之比,對化合物的基質效應狀況進行評價。基質效應按下式計算:
評價依據:80%≤基質效應≤120%,表示基質效應不明顯,可忽略不計;基質效應<80%或基質效應>120%,表示基質抑制或增強,說明存在基質效應。
1.4 數據處理
在TraceFinder 4.1分析軟件中,運用建立好的指紋識別數據庫對UPLC-Q/Orbitrap采集的樣品數據進行檢索匹配分析,利用質譜軟件的自動去卷積功能,并與指紋識別數據庫中的保留時間、精確母離子質量數、碎片離子質量數等相關參數進行對比(可疑化合物判定:保留時間偏差介于±0.5 min,離子質量數偏差介于±5×10-6), 針對可疑化合物利用HCD碎片離子譜圖庫進一步確證,使用基質匹配標準曲線外標法進行定量分析。借助OriginPro 2019b和WPS Office 2019搭配制圖。
2 結果與分析
2.1 前處理方法的優化
2.1.1 提取溶劑及體積的選擇
文獻[30-31]報道,常用提取劑有乙酸乙酯、乙腈、乙腈溶液且添加適量甲酸可有效增加提取效率,本實驗以多寶魚、鱖魚、烏魚和草魚為基質,以試樣加標測定值與試樣濃度之差除以加標量計算單個基質的平均加標回收率,考察乙酸乙酯、乙腈、體積分數80%與90%的乙腈溶液、含0.2%與1.0%甲酸的80%乙腈溶液6種提取劑下30種激素的提取效果,結果見圖1A。乙酸乙酯和純乙腈作為提取溶劑,4種魚肉的加標回收率處于較低水平,可能是兩者共提物較多且易結塊;乙腈溶液提取效率明顯高于乙酸乙酯和純乙腈,可能是乙睛溶液具有蛋白沉淀作用,其中體積分數80%的乙腈溶液提取效率高于體積分數90%,可能是高體積分數乙腈下目標化合物被大量變性蛋白質包裹而不易被解離提取;體積分數80%乙腈溶液中加入0.2%甲酸提取效率高于1.0%甲酸,可能是高體積分數甲酸降低了樣品溶解性,影響了離子化效率。故選擇0.2%甲酸-80%乙腈溶液為提取溶劑。
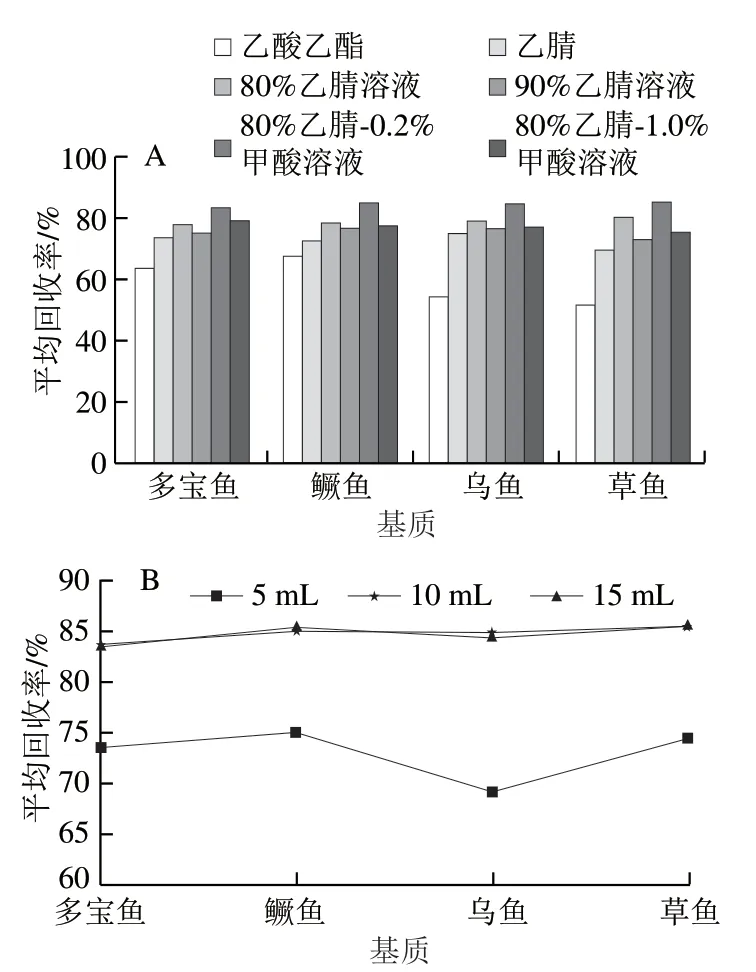
圖1 提取溶劑種類(A)及最適提取溶劑體積(B) 對30種激素回收率的影響Fig. 1 Effects of different extraction solvents (A) and different volumes of 80% acetonitrile aqueous solution containing 0.2% formic acid (B) on recoveries of 30 hormones
在此基礎上,實驗分別考察80%乙腈-0.2%甲酸溶液體積為5、10、15 mL對30種待測激素提取效果的影響,見 圖1B。使用10 mL提取溶劑的平均回收率均明顯高于5 mL,而10 mL與15 mL提取溶劑的平均回收率無顯著差異,可能是在10 mL提取溶劑下目標化合物已基本實現提取完全。考慮成本與環保節能,最終選擇10 mL作為提取劑體積。
2.1.2 固相萃取柱及填料量的選擇
魚肉基質復雜,樣品經提取后仍需凈化。實驗分別考察C18、Oasis HLB、Oasis PRiME HLB和Captiva EMR Lipid四種萃取柱對4 類魚肉中激素分離凈化效果,結果見圖2A。采用C18時,4 類魚肉中30種激素平均回收率最低,可能是由于傳統C18吸附劑難以同時兼顧所有激素性質,Oasis PRIME HLB的凈化效果均明顯優于Oasis HLB和Captiva EMR Lipid,表明Oasis PRIME HLB能夠更好地對樣品中的磷脂和蛋白質等雜質進行吸附。另有研究表明[32],PRIME HLB是多獸殘專用且適配高分辨質譜,該萃取柱無需活化與平衡且效果顯著,適合大規模樣品的快速凈化處理。
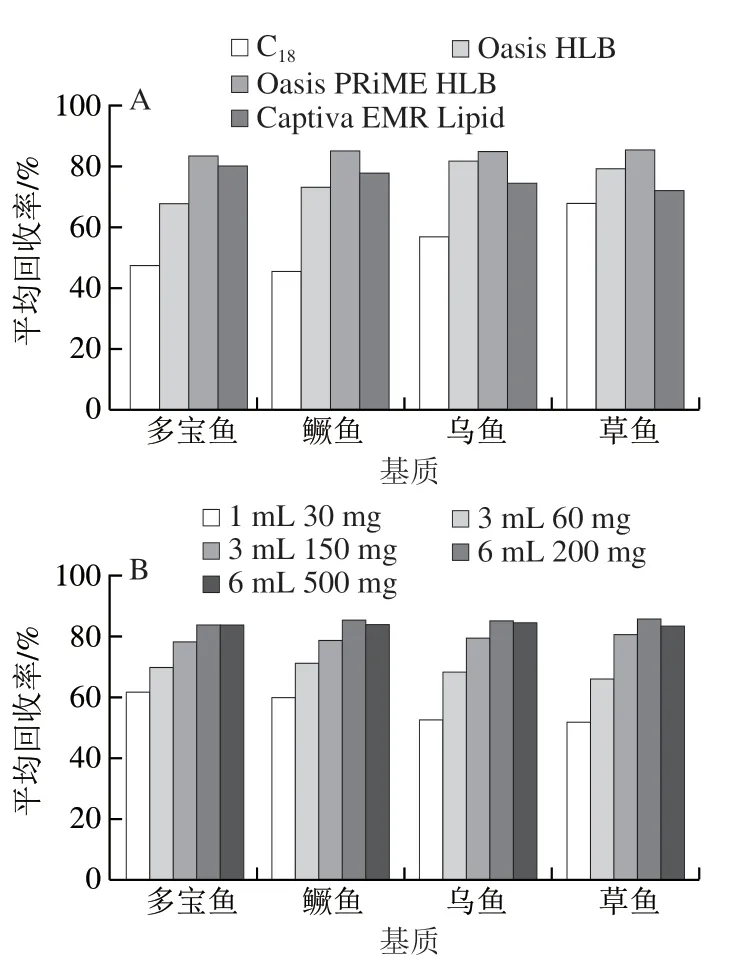
圖2 萃取柱(A)及填料量(B)對30種激素回收率的影響Fig. 2 Effects of different extraction columns (A) and packing amounts (B) on recoveries of 30 hormones
同時,實驗考察Oasis PRIME HLB不同填料用量(1 mL 30 mg、3 mL 60 mg、3 mL 150 mg、6 mL 200 mg、6 mL 500 mg)對目標物的吸附凈化效果。見圖2B。當填料由1 mL 30 mg增加到3 mL 150 mg時,4 類魚肉的萃取效率迅速提高;當填料高于3 mL 150 mg時效率增速放緩;當填料量為6 mL 200 mg和6 mL 500 mg時,兩者對多寶魚中激素的吸附凈化效果相當,但前者對其他3 類魚肉基質的吸附效果均最好。故最佳填料量為 6 mL 200 mg。
2.2 UPLC條件的優化
2.2.1 色譜柱的選擇
要同時實現對30種性質不同激素的有效分離,需要選取兼容性、柱效較好且耐用的色譜柱。實驗考察Thermo Accucore aQ C18(2.1 mm× 00 mm,2.6 μm)、Agilent Eclipse plus C18(3.0 mm×150 mm,1.8 μm)以及Waters Acquity BEH C18(2.1 mm×100 mm,1.7 μm)3種色譜柱對30種化合物的分離效果。結果表明,部分激素在Thermo Accucore aQ C18和Agilent Eclipse plus C18色譜柱上均存在不同程度的峰形拖尾和峰形尖銳度較差的現象。而Waters Acquity BEH C18色譜柱能夠較好保留30種激素化合物,峰形無明顯拖尾且尖銳對稱。可能是由于該色譜柱獨特的鍵合與極性基團端基封尾技術,確保了其兼顧不同極性化合物的保留性能更佳。故本實驗色譜柱選擇Waters Acquity BEH C18(2.1 mm× 100 mm,1.7 μm)。
2.2.2 流動相體系及流速的選擇
流動相體系直接影響分析物的保留時間和峰形,由于30種激素均易溶于乙腈,因此本實驗分別選取乙腈-水、乙腈-5 mmol/L乙酸銨、乙腈-20 mmol/L乙酸銨、乙腈-20 mmol/L乙酸銨(0.1%甲酸)共4種流動相組合進行分析。結果顯示:乙腈-水作為流動相時,各組分的峰形較差且正離子響應較低;乙腈-5 mmol/L乙酸銨所獲取的色譜峰形明顯優于乙腈-水,但部分蛋白同化激素存在峰形較差。當乙酸銨濃度加大至20 mmol/L時,峰形均得到明顯改善,繼續向乙腈-20 mmol/L乙酸銨溶液中添加0.1%甲酸,此時各待測物峰形均達到最佳,表明適量甲酸的加入有助于改善峰形。故本實驗流動相體系為乙腈-20 mmol/L乙酸銨(0.1%甲酸)溶液。在此基礎上考察0.3、0.4 mL/min和0.5 mL/min三種流速下待測物的分離效果。結果表明,在3種流速下,30種目標化合物均能有效分離,而當流速為0.5 mL/min時,不僅色譜峰形尖銳,還能夠最大限度地縮短保留時間,故流動相流 速為0.5 mL/min。
2.3 HRMS條件的優化
30種激素化合物在加熱電噴霧離子源下產生多種準分子碎片離子峰,實驗采用全掃描/數據依賴二級掃描(Full MS/dd-MS2)模式,對30種激素混合標準溶液進行正負離子同時掃描,得到一級全掃描質譜圖,以各化合物的準分子碎片離子峰理論精確質量數提取色譜圖,此條件下30種化合物的精確質量相對偏差均小于1.0×10-6,滿足實驗需求。同時,實驗還在多種分辨率值考察中發現,當一級全掃描質量分辨率為75 000,數據依賴二級掃描質量分辨率為17 500時,30種激素待測物與樣品基質中的干擾物均實現基線分離,響應值也得到明顯提高,表明在此分辨率條件下樣品中的基質干擾得到有效消減。圖3為30種激素的提取離子色譜圖。
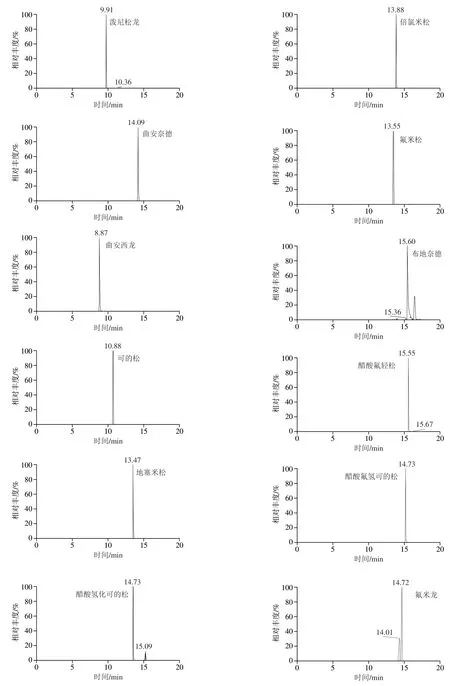
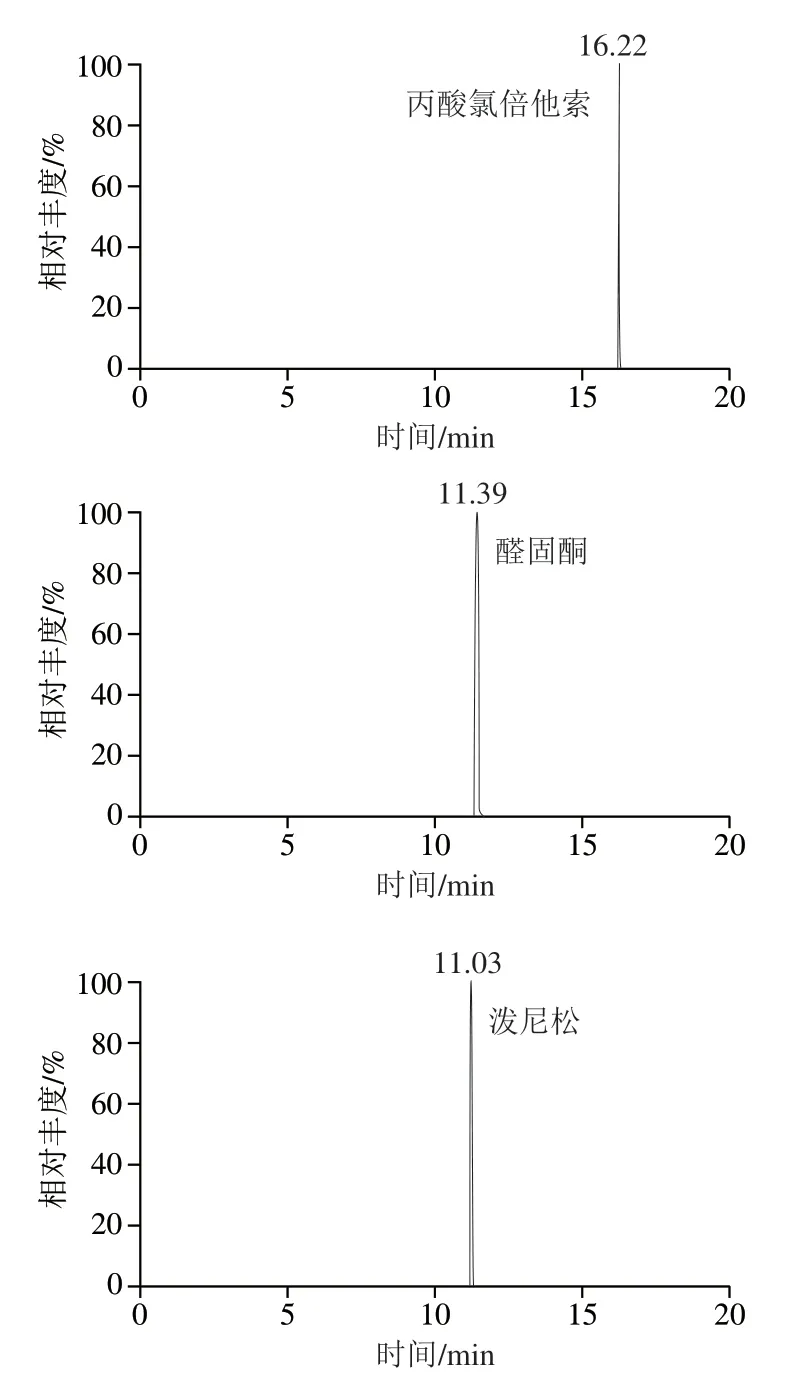
圖3 30種激素化合物的提取離子色譜圖Fig. 3 Extracted ion chromatograms of 30 hormone compounds
2.4 基質效應評價
魚肉基質復雜,可能存在基質抑制或基質增強效應,故進行基質效應評價。如表2所示,在多寶魚中, 30種激素的基質效應均在80%~120%范圍內,即基質效應不明顯;在鱖魚中,表睪酮、倍氯米松、潑尼松表現為基質抑制效應;在烏魚中,美雄酮、可的松表現為基質抑制效應,氫化可的松表現為基質增強效應;在草魚中,表睪酮、美雄酮、氟米松、氟米龍、潑尼松表現為基質抑制效應。為獲得更加準確的檢測結果,本實驗在定量環節通過基質匹配標準曲線消除或減弱基質效應的影響。
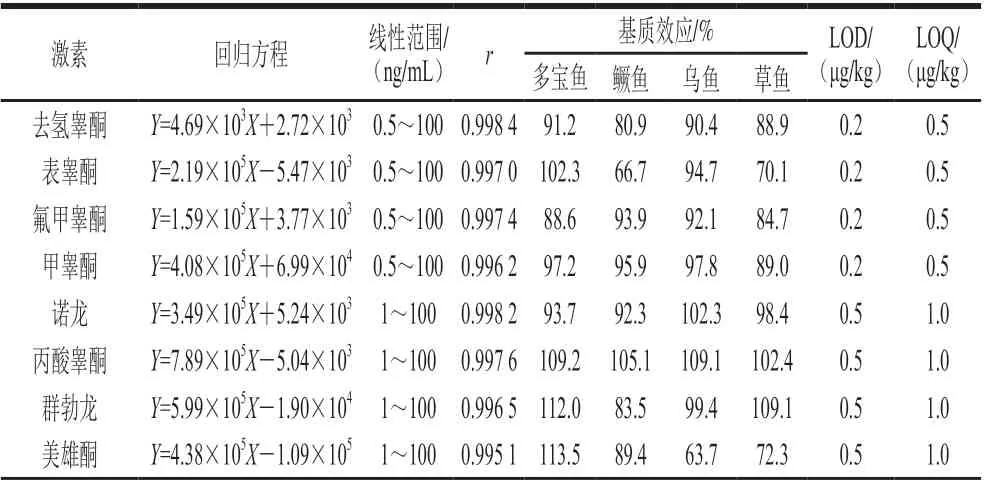
表2 30種激素的線性關系、基質效應、LOD和LOQTable 2 Linear relationships, matrix effects, limits of detection and limits of quantification of 30 hormones
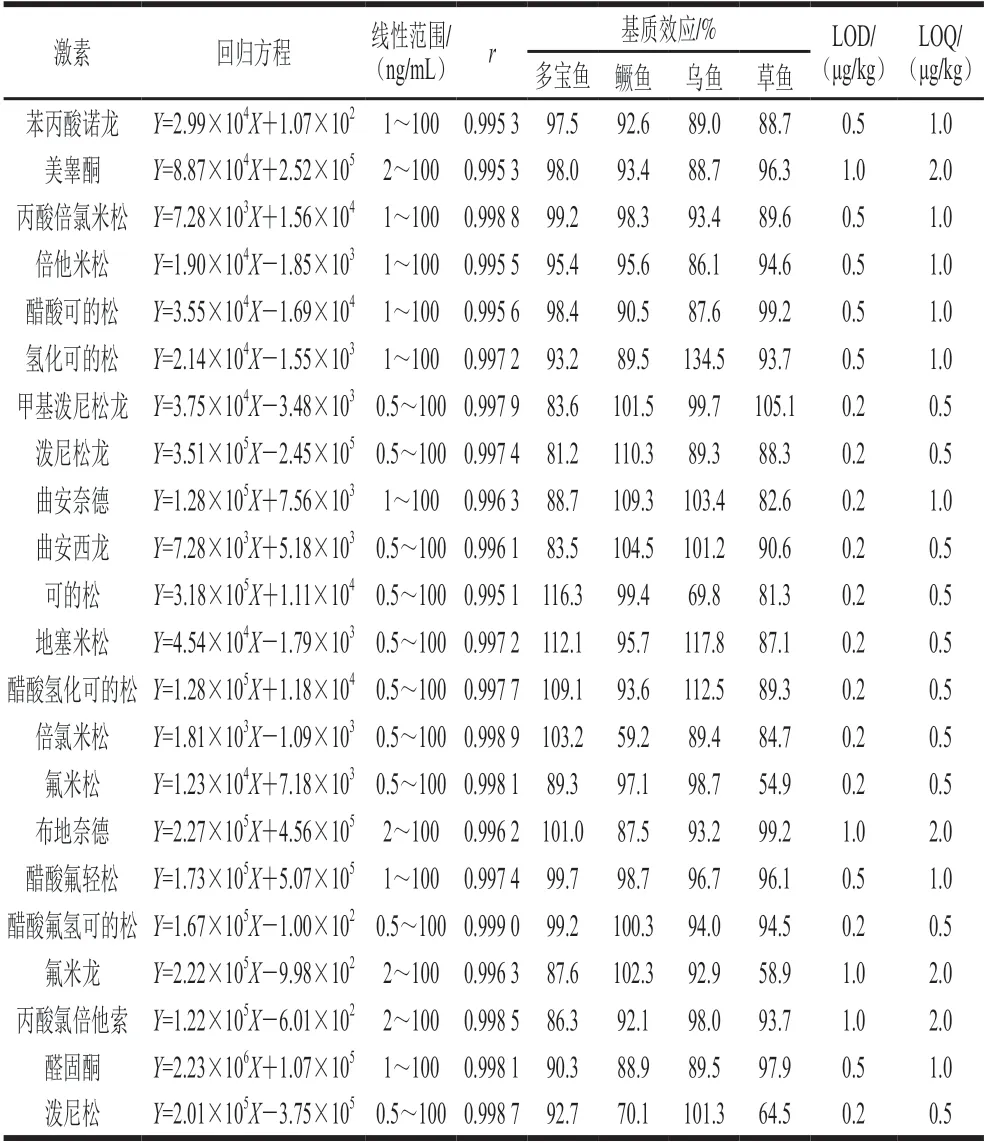
續表2
2.5 方法的線性關系、基質效應、LOD和LOQ
將魚肉空白提取液配制30種化合物的標準工作液,在已優化的分析條件下進行測定。以目標化合物色譜峰面積(Y)及其對應組分質量濃度(X)繪制標準曲線,并計算獲得待測物質的線性回歸方程及其相關系數(r),分別以3 倍信噪比和10 倍信噪比計算出方法的LOD和LOQ。由表2可知,魚肉中30種激素在0.5~100 ng/mL質量濃度范圍內線性關系良好,相關系數(r)均大于0.995 0。方法的LOD和LOQ依次為0.2~1.0 μg/kg和0.5~2.0 μg/kg,均滿足實驗需求。
2.6 回收率和精密度結果
向多寶魚、鱖魚、烏魚、草魚的空白樣品中分別添加30種激素的標準溶液配制成陽性樣品,按照已建立的方法,在1、5 倍和10 倍LOQ三個添加水平下各重復測定6 次,求解得到回收率及相對標準偏差(relative standard deviation,RSD),結果見表3。添加水平在1~10 倍LOQ時,30種激素在多寶魚基質中的回收率為72.1%~103.2%,RSD為2.6%~9.4%;在鱖魚基質中的回收率為69.7%~101.1%,RSD為2.3%~8.9%;在烏魚基質中的回收率為69.9%~99.3%,RSD為3.2%~9.2%;在草魚基質中的回收率為69.9%~103.2%,RSD為2.8%~9.1%。表明本研究建立的方法準確度和精密度均滿足實驗需求。

表3 30種激素在魚肉中的回收率和RSD(n=6)Table 3 Recoveries and RSDs of 30 hormones spiked in fish (n = 6)%
2.7 實際樣品篩查
應用本研究建立的方法對36 批次市售魚肉樣品(多寶魚、鱖魚、烏魚和草魚各9 批次)進行檢測,樣品經前處理和上機測試后,利用TraceFinder軟件中已建數據庫對樣品中30種激素進行多目標定性篩查,可疑樣品結合HCD二級特征碎片離子確證,使用基質匹配標準曲線外標法定量。1 批次樣品草魚中檢出氫化可的松 (3.9 μg/kg),其余樣品均未檢出30種激素藥物。各項指標均達到篩檢要求,表明結果準確可靠。
3 結 論
本實驗以4種魚肉作為研究對象,建立多目標快速篩查和確證魚肉中30種蛋白同化激素及糖皮質激素的分析方法,樣品經80%乙腈-0.2%甲酸溶液提取后離心,上清液經Oasis PRiME HLB固相萃取柱凈化后過濾膜上機測試。30種激素的添加回收率為69.7%~103.2%,RSD為2.3%~9.4%;LOD為0.2~1.0 μg/kg,LOQ為0.5~2.0 μg/kg。 該方法優勢明顯,更加快捷高效、準確可靠,能滿足日常魚肉樣品中30種蛋白同化激素及糖皮質激素快速篩查的需要。
