動物源性食品中硝基酚鈉殘留量的測定
2022-03-09 01:49:16徐瑞麗李豐袁曉劉姚紅楊悠楊昌彪
食品工業 2022年2期
徐瑞麗,李豐,袁曉,劉姚紅,楊悠,楊昌彪
1. 廣電計量檢測(湖南)有限公司(長沙 410007);2. 貴州省分析測試研究院(貴陽 550002)
復方硝基酚鈉(復硝酚鈉、硝基酚鈉)是由5-硝基鄰甲氧基苯酚鈉、鄰硝基苯酚鈉和對硝基苯酚鈉按照一定比例調配而成的生長調節劑。由于硝基酚鈉高效低毒,具有促根壯苗、提高產量、提高產品品質等作用[1-7],因此作為植物生長調節劑而被廣泛應用。同時,硝基酚鈉也可作為飼料添加劑,用于提高肉、蛋、毛、皮等產品的產量和質量,并可預防多種疾病[8]。研究表明,硝基酚鈉為弱致敏物質[9],雖然其毒性較低[10],但對人類健康仍具有潛在危害。2002年我國先后在中華人民共和國農業部公告第193號《食品動物禁用的獸藥及其他化合物清單》和中華人民共和國農業部公告第235號《動物性食品中獸藥最高殘留限量》中將硝基酚鈉列為所有食品動物中的禁用藥物(2019年12月27日農業農村部將禁用藥物相關公告修訂為中華人民共和國農業農村部公告第250號《食品動物中禁止使用的藥品及其他化合物清單》)。
硝基酚鈉的檢測方法主要為分光光度法[11]、液相色譜法[12-13]、氣相色譜法[14-16]、液相色譜串聯質譜法[17-19]等。其中,分光光度法干擾較大,靈敏度較低;液相色譜法靈敏度低,而且主要為原藥及飼料肥料的檢測[20-22],氣相色譜法需將目標物進行衍生后測定,方法操作較繁瑣。關于動物源性食品中硝基酚鈉殘留檢測的研究較少[13,16,23-24],而使用固相萃取-高相液相色譜串聯質譜法檢測動物源性食品中硝基酚鈉的檢測方法尚未見報道。為此,試驗開發一種使用乙腈超聲提取,經陰離子固相萃取柱凈化,C18色譜柱分離,超高效液相色譜串聯質譜法檢測動物源性食品中硝基酚鈉殘留量的測定方法。
1 材料與方法
1.1 材料與試劑
5-硝基鄰甲氧基苯酚鈉、鄰硝基苯酚鈉和對硝基苯酚鈉(純度大于99%;Be Pure);甲醇、乙腈(色譜純,上海安譜實驗科技股份有限公司);甲酸(色譜純,天津市科密歐化學試劑有限公司);濃氨水(25%~28%,西隴科學股份有限公司);試驗用水為一級水。陰離子固相萃取小柱(MAX,60 mg/3 mL;北京中檢維康技術有限公司)。
1.2 儀器與設備
超高效液相色譜串聯質譜儀(UPLC-MS/MS):(Xevo TQ-S Micro,配有電噴霧離子源,美國沃特世公司);渦旋混勻器(VORTEX-6,其林貝爾儀器制造有限公司);超聲波清洗器(AS30600BT,廣州利橋儀器科技有限公司);低速離心機(LD5-2B,廣州市丹安儀器儀表有限公司);高速離心機(H1850,湖南湘儀實驗室儀器開發有限公司)。
1.3 標準溶液配制
1.3.1 1 .0 mg/mL標準儲備液
分別稱取10 mg(精確到0.01 mg)5-硝基鄰甲氧基苯酚鈉、鄰硝基苯酚鈉和對硝基苯酚鈉標準物質,用甲醇溶解并定容到10 mL。于4 ℃冰箱中冷藏保存。
1.3.2 混合標準中間溶液
用甲醇將對硝基苯酚鈉、5-硝基鄰甲氧基苯酚鈉和鄰硝基苯酚鈉標準儲備液稀釋為質量濃度分別為1.0,1.0和100 mg/L的混合標準中間溶液,于4 ℃冰箱中冷藏保存。
1.3.3 混合標準工作溶液
根據需要,臨用前吸取一定量的混合標準中間溶液,用2%甲酸甲醇-水(3∶2)溶液配制成適當濃度的混合標準工作溶液。
1.4 樣品前處理
1.4.1 提取
稱取5.0 g(精確到0.001 g)樣品于50 mL離心管中,加入10 mL乙腈,渦旋混勻并超聲提取10 min。以4 000 r/min的轉速離心5 min,取上清液于另一離心管中。用10 mL乙腈重復提取1次,合并上清液。提取液中加入4 mL氨水,渦旋混勻30 s。以8 000 r/min高速離心5 min,取上清液待凈化。
1.4.2 凈化
取MAX固相萃取柱置于固相萃取儀,先后用3 mL甲醇和3 mL水活化,棄去流出液。將提取液轉入固相萃取小柱,以每秒1~2滴的流速使樣品通過小柱,棄去流出液。依次用3 mL 5%氨水溶液,3 mL甲醇淋洗。用3 mL 2%甲酸甲醇溶液洗脫并收集流出液。洗脫液用水定容至5 mL,渦旋混勻,過濾膜后上機測定。
1.5 儀器條件
1.5.1 液相條件
色譜柱,ACQUITY UPLC? BEH C18柱(1.7 μm,2.1 mm×50 mm);流速0.3 mL/min;進樣量10 μL;柱溫35 ℃。流動相:A為5 mmol/L乙酸銨溶液,B為甲醇。梯度洗脫程序:0~2 min,90% A,2~4 min,90%~60% A;4~4.5 min,60%~10% A;4.5~7 min,10% A;7~8 min,10%~90% A;8~10 min,90% A。
1.5.2 質譜條件
離子源,電噴霧離子源(ESI);掃描模式,負離子掃描;監測方式,多反應監測(MRM);毛細管電壓3.0 kV;離子源溫度150 ℃;脫溶劑氣溫度500 ℃,脫溶劑氣流量1 000 L/h;碰撞氣流量50 L/h;3種目標物的保留時間、母離子、子離子、去簇電壓、碰撞電壓等參數見表1。

表1 對硝基苯酚鈉、5-硝基鄰甲氧基苯酚鈉和鄰硝基苯酚鈉的質譜參數
2 結果與討論
2.1 色譜條件優化
2.1.1 質譜條件的優化
在負離子模式下,對3種目標物分別進行母離子和子離子掃描,確定高響應特征離子對。分別優化3種目標物的毛細管電壓、去簇電壓、碰撞電壓等參數,確定最佳儀器條件,詳細參數見表1。
2.1.2 液相條件的優化
對比在使用不同比例的甲醇-水、乙腈-水、甲醇-甲酸水溶液、乙腈-甲酸水溶液、甲醇-乙酸銨溶液、乙腈-乙酸銨溶液、甲醇-0.1%氨水溶液等不同體系的流動相時3種目標物的響應及分離效果。結果發現,在使用甲醇-水、乙腈-水、甲醇-甲酸水溶液、乙腈-甲酸水溶液作為流動相時,鄰硝基酚鈉響應特別低。使用甲醇-5 mmol/L乙酸銨溶液作為流動相時,鄰硝基酚鈉響應明顯增強,3種目標物峰形良好且分離度較好,3種目標物的總離子流(TIC)圖見圖1。
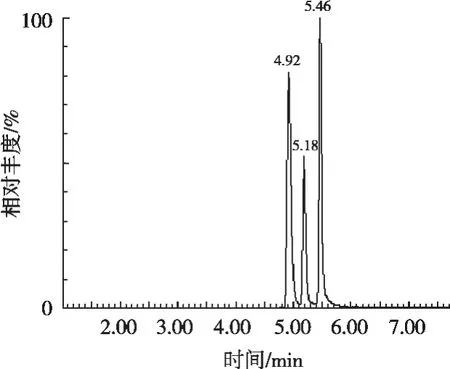
圖1 混合標準溶液總離子流圖
2.2 前處理條件優化
2.2.1 提取溶劑
硝基苯酚鈉為強極性化合物,易溶于水,可溶于甲醇、乙醇、乙腈、丙酮等有機溶劑。對比甲醇、乙腈、乙腈-水、氨水乙腈等不同溶劑的提取效果。結果發現,使用乙腈作為提取溶劑時,3種目標物的提取效率最高。同時乙腈具有沉淀蛋白的作用,使用乙腈提取時,提取液較澄清易于離心分離,因此選擇乙腈作為提取溶劑。在提取完成后,在提取液中加入適量氨水,使3種目標物均以陰離子形式存在。在提取液上柱時,可增強目標物與陰離子固相萃取柱中的陽離子基團的離子間作用力,從而增強目標物在小柱上的保留。
2.2.2 凈化方式
5-硝基鄰甲氧基苯酚鈉、鄰硝基苯酚鈉和對硝基苯酚鈉以陰離子形式存在時,可使用陰離子固相萃取柱對提取液進行凈化和富集。
2.2.3 濃縮及定容
鄰硝基苯酚鈉在濃縮時條件比較苛刻,濃縮過程較難控制;同時濃縮操作會增強樣品的基質效應,降低目標物的響應。因此選擇提取液經小柱凈化后,洗脫液不進行濃縮操作。同時在洗脫液中加入適量的水,以降低目標物的基質效應及樣品在儀器分析時的溶劑效應。
2.3 檢出限與線性范圍
用2%甲酸甲醇-水(3∶2)溶液配制對硝基酚鈉和5-硝基鄰甲氧基苯酚鈉質量濃度分別為0.5,1.0,2.0,5.0,8.0和10.0 μg/L,鄰硝基酚鈉質量濃度分別50,100,200,500,800和1 000 μg/L的混合標準工作溶液后上機檢測。結果發現,在對硝基酚鈉和5-硝基鄰甲氧基苯酚鈉在0.5~10.0 μg/L質量濃度范圍內,鄰硝基酚鈉在50~1 000 μg/L質量濃度范圍內,峰面積y與分析物的質量濃度x(μg/L)均具有良好的線性關系,相關系數R2均大于0.999,滿足方法學要求。通過空白樣品添加目標物的方法,在信噪比為3時確定3種組分的檢出限。3種組分的線性回歸方程、線性范圍、線性系數及檢出限見表2。
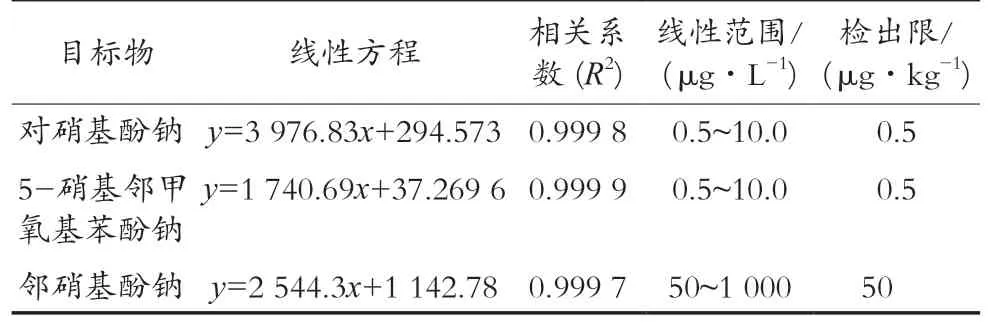
表2 工作曲線方程、相關系數、線性范圍及檢出限
2.4 回收率與精密度
對于禁用物質,分別選擇檢出限、2倍檢出限和10倍檢出限3個濃度水平進行回收率和精密度試驗。分別選擇豬肉(畜肉)、雞肉(禽肉)、魚肉(水產品)3種空白樣品作為動物源性食品代表基質,進行添加回收試驗。試驗結果表明對硝基酚鈉和5-硝基鄰甲氧基苯酚鈉在0.5,1.0和5.0 μg/kg 3個添加水平的平均回收率分別為75.3%~93.9%和71.9%~93.6%,相對標準偏差分別為3.3%~14%和3.3%~5%,鄰硝基酚鈉在50,100和500 μg/kg 3個不同質量分數添加水平的平均回收率在64.1%~91.1%,相對標準偏差為2.8%~ 15%,詳見表3。結果表明,該方法的準確度和精密度均可滿足食品中藥物殘留分析的要求。

表3 回收率與精密度試驗結果(n=6)
3 結論
建立用乙腈超聲提取,陰離子固相萃取柱凈化,C18色譜柱分離,超高效液相色譜串聯質譜法檢測動物源性食品中復方硝基酚鈉3種組分殘留量的分析方法。結果表明,對硝基酚鈉和5-硝基鄰甲氧基苯酚鈉在0.5~10.0 μg/L、鄰硝基酚鈉在50~1 000 μg/L質量濃度范圍內線性關系良好,線性相關系數均大于0.999,對硝基酚鈉和5-硝基鄰甲氧基苯酚鈉檢出限為0.5 μg/kg,鄰硝基酚鈉檢出限為50 μg/kg。對硝基酚鈉和5-硝基鄰甲氧基苯酚鈉在0.5,1.0和5.0 μg/kg 3個添加水平的平均回收率分別為75.3%~93.9%和71.9%~93.6%,相對標準偏差分別為3.3%~14%和3.3%~15%,鄰硝基酚鈉在50,100和500 μg/kg 3個不同質量分數添加水平的平均回收率在64.1%~91.1%,相對標準偏差為2.8%~15%。該方法操作簡便,回收率和精密度較好,準確度高,可用于動物源性食品中復方硝基酚鈉殘留量的檢測。
