基于CiteSpace的肌肉萎縮側索硬化研究可視化分析
2022-04-08 03:38:58劉歡慶王林峰
醫學信息 2022年6期
關鍵詞:研究
劉歡慶,王林峰
(1.長沙縣人民醫院/湖南省人民醫院星沙院區電生理科,湖南 長沙 410004;2.湖南中醫藥大學信息科學與工程學院,湖南 長沙 410208)
肌肉萎縮側索硬化(amyotrophic lateral sclerosis,ALS)是一種進行性神經系統變性疾病,主要累及患者大腦皮層、腦干和脊髓運動神經元[1,2]。該病以進行性加重的骨骼肌無力、萎縮、肌束顫動、延髓麻痹和錐體束征為主要臨床表現,一般好發于中老年人群[3]。目前該病尚無有效的治療方法,生存期通常為3~5年,然而隨著研究人員的不斷努力,該疾病癥狀前期的病理生理特點及發病機制逐漸熟知,這為制定更有效的治療方案提供了啟示。為了更好地了解該疾病的研究熱點與前沿動態,本研究采用陳超美教授開發的文獻知識圖譜分析軟件CiteSpace V 對國內近20年肌萎縮側索硬化相關研究進行可視化分析,旨在為該疾病的研究提供更多支撐信息。
1 資料與方法
1.1 數據來源 本研究的數據均來源于中國知網數據庫,在專業檢索模式下進行文獻檢索,檢索表達式為:ti=肌肉萎縮側索硬化+肌萎縮側索硬化+漸凍癥-進展-現狀-概況-概述-Meta 分析。檢索時段為2000年1月1 日-2020年12月31 日。
1.2 納入與排除標準 納入標準:研究主題為肌肉萎縮側索硬化的案例報導、臨床試驗、實驗研究、名醫經驗等。由3 名研究人員分別逐一閱讀文獻摘要進行文獻納入與排除,意見不一致時討論解決,若未達成共識則投票決定。排除標準:①與肌肉萎縮側索硬化主題無關的文獻;②診療指南;③會議、報紙、心得體會及未標明作者的文獻;④綜述、Meta 分析等其他類型文獻;⑤重復文獻。
1.3 方法 將篩選后的文獻以Refworks 格式導出,導出的文獻信息包括標題、摘要、關鍵詞、作者及研究機構等,導出的txt 文件以download_格式命名后導入至CiteSpace 5.7.R2 中進行數據轉換,基于轉換后的數據文件,設置運行參數:年份為2000-2020年,Years Per Slice 為1,Top N 為50,剪裁參數:無。
2 結果
2.1 檢索結果 基于以上檢索表達式,共檢索到文獻1304 篇,根據排除標準共排除278 篇文獻,最終納入1026 篇文獻。
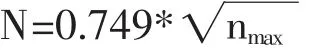
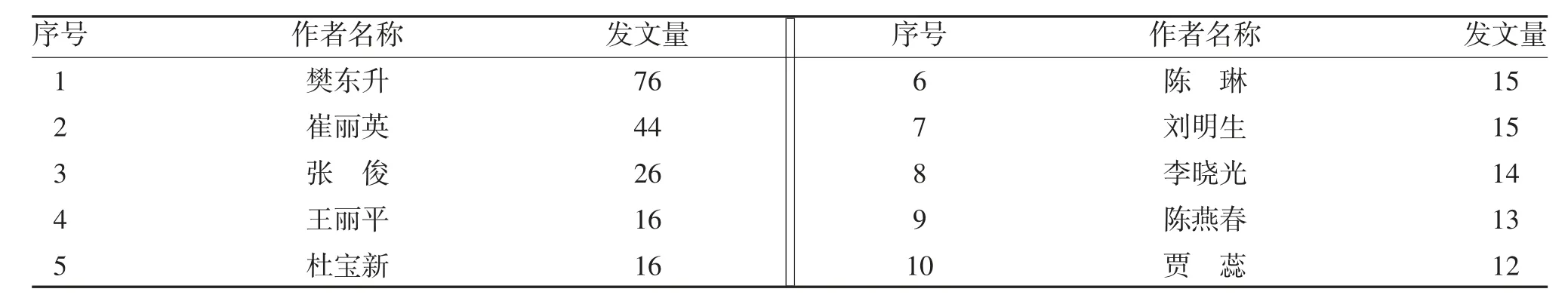
表1 發文量排名前10的作者
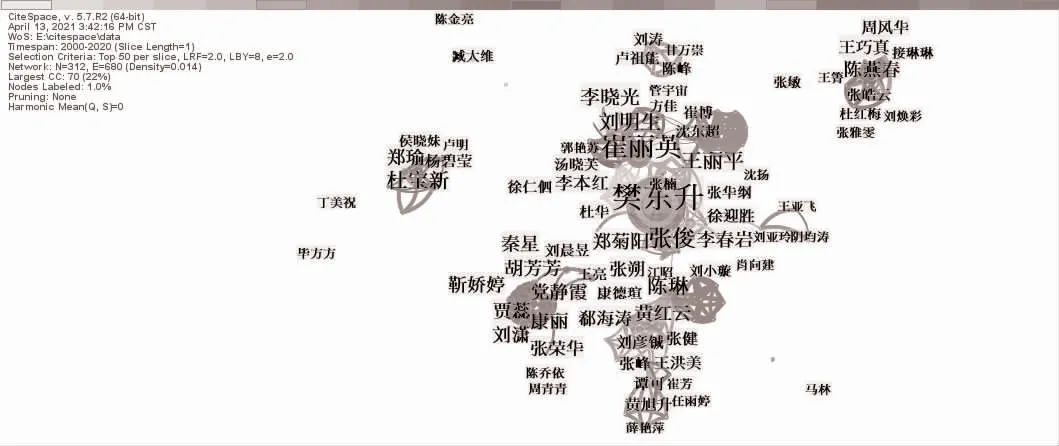
圖1 作者合作網絡
2.3 機構分析 運行CiteSpace 軟件,節點類型選擇institution,得到N=83,E=48的單位合作網絡圖譜見圖2,表明肌肉萎縮側索硬化的研究機構共83個,其中發文量排在前10 位的機構見表2。機構之間以北京大學第三醫院神經內科、中國醫學科學院北京協和醫院神經內科為中心組成了較小的合作網絡。節點形狀越大表示發文量越多,由此說明北京大學第三醫院神經內科在該領域處于核心地位,學術科研力量強。圖中節點之間的連線數較少,且Denisty 參數為0.0123<0.02,說明各機構之間的合作關系不強。
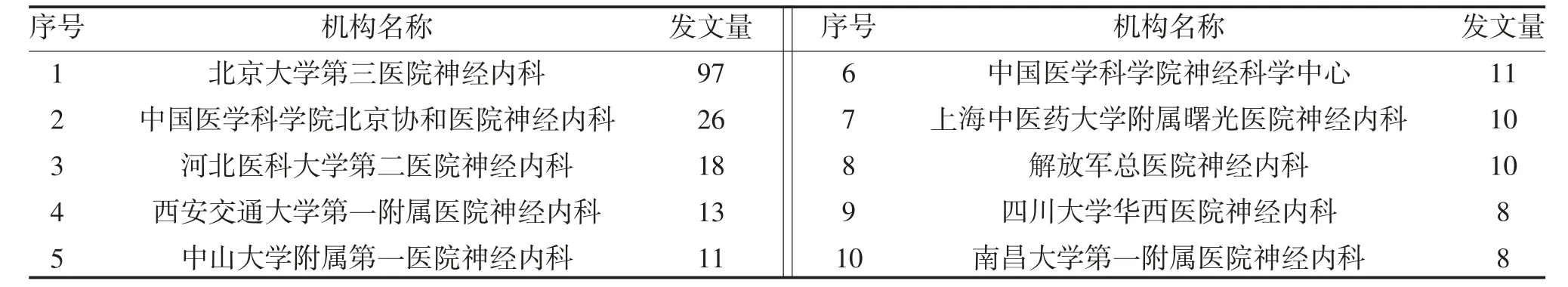
表2 發文量排名前10的機構

圖2 機構合作圖譜
2.4 關鍵詞分析
2.4.1 關鍵詞共現 運行CiteSpace 軟件,節點類型選擇keyword,得到N=108,E=80的關鍵詞共現圖譜見圖3。通過對同義關鍵詞進行合并后,頻次排名前10的關鍵詞為:肌電圖(47),護理(20),基因突變(16),診斷(15),預后(12),錐體束(11),自噬(11),谷氨酸(11),頸椎病(10),神經傳導(9)。

圖3 關鍵詞共現圖譜
2.4.2 關鍵詞聚類 在關鍵詞共現網絡基礎上,采用軟件推薦使用的LLR 算法對關鍵詞進行聚類分析,得到模塊化Q 值(Modularity Q)=0.6556,平均輪廓值(Weighted Mean Silhouette S)=0.8792的關鍵詞聚類圖譜見圖4。該圖譜劃分了10 個聚類,分別為:#0 內質網應激、#1 小鼠、#2 肌電圖、#3 磁共振成像、#4 包涵體、#5 診斷、#6 丙二醛、#7 基因多態性。采用Clusters 菜單下的Summary Table 功能得到聚類主題匯總表見表3,該表只列出每個聚類的前5 個主題。

表3 聚類主題匯總表
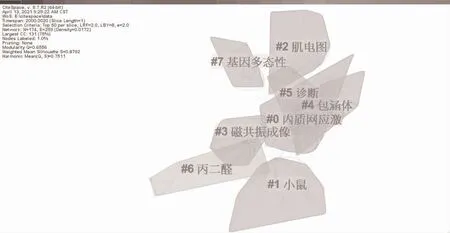
圖4 關鍵詞聚類圖譜
2.4.3 關鍵詞突現 在關鍵詞共現分析基礎上,通過運行CiteSpace 軟件的Burstness 功能,共得到21 個關鍵詞,見圖5。

圖5 關鍵詞突現
3 討論
通過關鍵詞共現、聚類、突現可視化分析可總結出國內對于ALS的研究熱點和趨勢可大致分為兩個階段。第一個階段為2000-2010年,研究主題主要集中在臨床表現與診斷方面。第二個階段為2011-2020年,研究主題主要集中在生物學層面,在該階段的早期(2011-2015年),研究主題主要集中在細胞層面,如內質網應激、線粒體功能障礙;2015年后研究主題主要在分子層面,如血紅素氧合酶1(haem oxygenases-1,HO-1)、tdp-43 結合蛋白、C9orf72 基因等。
3.1 2000-2010年階段在早期階段,ALS的確診主要依靠臨床表現[5],但其臨床癥狀與其他一些神經系統疾病相同或類似,容易誤診[6,7]。在肌電圖異常納入ALS的ElEscorical 診斷標準中后,探討ALS的臨床表現和肌電圖特征成為該階段的研究熱點[8]。四肢遠近端肌肉在靜息狀態下出現自發電位(如纖顫電位、正銳波、束顫電位)及在輕收縮時出現寬時限、高波幅的運動單位電位,特別是針極肌電圖MUP 出現,提示脊髓前角細胞受損的巨大電位有助于ALS疑似患者確診,但這些不是ALS 特有的電生理表現。舌肌、面肌、胸鎖乳突肌、椎旁肌等肌肉的針極肌電圖與ALS的臨床特征高度相關,其中以顱神經支配的舌肌異常針極為ALS的確診提供了重要證據。在臨床診治過程中,肌電圖檢查不僅能輔助確診ALS,還有助于評估疾病的累及范圍、進展速度、嚴重程度及臨床治療的效果。
3.2 2011-2020年階段隨著分子生物技術的不斷發展,越來越多的致病機理被發現,如線粒體功能障礙、蛋白質異常聚集、自噬-溶酶體功能紊亂等[9,10]。近10年來國內對該疾病的研究主題也主要集中在致病機理的研究方面。由圖5 可知,自2011年開始,內質網應激、自噬、HO-1、氧化應激、C9ORF72 等因素陸續被發現,然而這些關鍵詞的突現年限(開始年份至截止年份)并不長,這間接說明ALS的致病因素較多且復雜。由表3 中聚類#7 出現“基因多態性”“散發性肌肉萎縮側索硬化”等標簽詞可知,基因多態性或許是造成ALS 發病機制較復雜的重要原因。有研究表明在我國的ALS 人群中,SOD1 基因突變達20 多種[11]。除了tdp-43、C9ORF72 等基因外,還有更多突變基因是散發性ALS 患者的候選致病基因,國外的相關研究也證實了這點[12-14]。
針對基因突變導致的ALS,基因治療可能會是一種有效的治療方法。例如腺相關病毒的載體可以有效地靶向基因,借助病毒載體可以將反義寡核苷酸導入中樞神經系統,以穩定地轉導中樞神經系統中的運動神經元[15]。這種治療方法在德國ALS 患者SOD1 和C9ORF72 基因突變的臨床試驗中獲得了良好的結果[16]。雖然有較多的致病基因被揭曉,但該疾病的致病基因可能存在一定的種族差異性。國外有研究顯示[17,18],ALS 潛在候選致病基因如TUBA4A、TBK1 等,不一定是國內ALS 患者的主要致病基因。因此,更多國內ALS 患者的候選致病基因仍有待進一步深入研究。
4 總結
目前國內ALS 領域以樊東升、崔麗英為代表的團隊處于主導地位,在該領域作出了重要貢獻。近幾年也誕生了少數新的研究團隊,但能否持續性研究值得關注。機構分析表明,作者間的合作關系大部分限于機構內部作者之間的合作,機構之間的合作關系非常薄弱。研究主題方面,從早期的臨床表現與肌電圖特征向分子生物層面致病機制研究深入,未來基因治療可能會成為ALS 安全有效的治療方式。雖然本研究未納入Web of Science 平臺中的英文文獻,但是本研究旨在通過對國內學者圍繞ALS 相關研究進行分析,為該領域的研究提供有價值的參考信息。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
遼金歷史與考古(2019年0期)2020-01-06 07:45:20
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年11期)2018-08-04 03:26:04
汽車工程學報(2017年2期)2017-07-05 08:13:02
國際商務財會(2017年8期)2017-06-21 06:14:14
電子制作(2017年23期)2017-02-02 07:17:19
